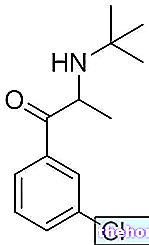
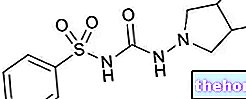
Активни састојци: Сунитиниб
СУТЕНТ 12,5 мг тврде капсуле
СУТЕНТ 25 мг тврде капсуле
СУТЕНТ 37,5 мг тврде капсуле
СУТЕНТ 50 мг тврде капсуле
Индикације Зашто се Сутент користи? За шта је то?
Сутент садржи активну супстанцу сунитиниб, која је инхибитор протеинске киназе. Користи се за лечење рака спречавањем активности одређене групе протеина за које је познато да су укључени у раст и ширење ћелија рака.
Сутент ће вам прописати само лекар који има искуства у употреби лекова против рака.
Сутент се користи за лечење одраслих са следећим врстама рака:
- Гастроинтестинални стромални карцином (ГИСТ), врста рака желуца и црева, у случајевима када иматиниб (други лек против рака) више не делује или се више не може узимати.
- Метастатски рак бубрега (МРЦЦ), врста рака бубрега која се проширила на друге делове тела.
- Неуроендокрини тумори панкреаса (пНЕТ) (тумори ћелија панкреаса који производе хормоне) који напредују или се не могу ресектовати
. Ако нисте сигурни како Сутент делује или зашто вам је прописан овај лек, питајте свог лекара.
Контраиндикације Када се Сутент не сме користити
Не узимајте Сутент:
- Ако сте алергични на сунитиниб или неки други састојак овог лека (наведен у одељку 6).
Мере опреза при употреби Шта треба да знате пре него што узмете лек Сутент
Реците свом лекару пре него што узмете Сутент:
- Ако имате висок крвни притисак. Сутент може узроковати пораст крвног притиска. Ваш лекар може проверити ваш крвни притисак док узимате Сутент и мораће да узима лекове за снижавање крвног притиска ако је потребно.
- Ако имате или сте имали поремећаје крви, проблеме са крварењем или модрице. Лечење Сутентом може носити повећани ризик од крварења, промене у броју одређених крвних зрнаца чији недостатак доводи до анемије или утиче на способност згрушавања крви.Ризик од крварења може бити већи ако узимате варфарин или аценокумарол, лекове који разређују крв како би спречили настанак крвних угрушака. Реците свом лекару ако доживите крварење током узимања лека Сутент.
- Ако имате проблема са срцем. Сутент може изазвати проблеме са срцем. Реците свом лекару ако се осећате јако уморно, имате недостатак ваздуха или имате отечена стопала и глежњеве.
- Ако доживите абнормалне промене срчаног ритма. Сутент може изазвати промене у срчаном ритму. Док се лечите Сутентом, Ваш лекар ће можда урадити електрокардиограм да процени обим ових промена. Реците свом лекару ако осетите вртоглавицу, несвестицу или имате абнормалне откуцаје срца док узимате Сутент.
- Ако сте недавно имали проблема са крвним угрушцима у венама и / или артеријама (врсте крвних судова), укључујући мождани удар, срчани удар, емболију или тромбозу. Одмах се обратите лекару ако осетите симптоме као што су стезање или бол у грудима, бол у рукама, леђима, врату или вилици, отежано дисање, утрнулост или слабост на једној страни тела, дрхтаво ходање, бол током лечења Сутентом. Главобоља или вртоглавица.
- Ако имате проблема са штитном жлездом. Сутент може изазвати проблеме са штитном жлездом. Реците свом лекару ако се лакше уморите док узимате Сутент, генерално вам је хладније од других људи или вам се глас смањи. Функцију штитне жлезде треба проверавати пре узимања лека Сутент и редовно током узимања лека. Ако штитна жлезда не производи довољно хормона штитне жлезде, можда ће бити потребно да узмете заменски хормон штитне жлезде.
- Ако имате или сте имали проблема са панкреасом или жучном кесом. Реците свом лекару ако приметите било који од следећих знакова и симптома: бол у стомаку (горњем делу стомака), мучнина, повраћање и грозница. То може бити узроковано упалом панкреаса или жучне кесе.
- Ако имате или сте икада имали проблема са јетром. Реците свом лекару ако осетите било који од следећих знакова и симптома проблема са јетром током лечења Сутентом: свраб, жутило коже или очију, тамни урин и бол или нелагодност у горњем десном делу стомака. Ваш лекар би требао спровести тестове како би се проверила функција јетре пре и током лечења Сутентом, и према клиничкој потреби.
- Ако имате или сте имали проблема са бубрезима. Лекар ће пратити функцију бубрега.
- Ако вам предстоји операција или сте недавно имали операцију. Сутент може утицати на начин на који зарастају ваше ране. Уопштено говорећи, ако вам предстоји операција, мораћете да престанете да користите Сутент. Ваш лекар ће одлучити када ће поново започети Сутентно лечење.
- Пре почетка лечења Сутентом препоручљиво је проћи стоматолошки преглед.
- ако имате или сте имали бол у устима, зубима и / или вилици, оток или ране у устима, утрнулост или осећај тежине у вилици или отпуштање зуба, одмах обавестите свог лекара и стоматолога.
- ако се подвргавате инвазивном зубном лечењу или зубној операцији, реците свом лекару да се лечите Сутентом, нарочито ако узимате и интравенозне бисфосфонате или сте их претходно узимали. Бисфосфонати су лекови који се користе за спречавање коштаних компликација које су можда прописане за неки други медицински проблем.
- Ако имате или сте икада имали поремећаје коже и поткожног ткива. "Гангренозна пиодерма" (болно улцерисање коже) или "некротизирајући фасциитис" (брзо ширења "инфекције коже / меког ткива која може бити фатална) могу се јавити током лечења овим леком. Прекид лечења. Озбиљне кожне реакције (Стевенс-Јохнсон синдром, токсична епидермална некролиза, мултиформни еритем) пријављени су уз употребу сунитиниба, који се у почетку појављивао на трупу као црвенкасте мрље у облику мете или кружне мрље, често са жуљевима у средини. Реакција може напредовати до раширених пликова или љуштења коже, а може бити и фатална. Ако се појави осип или неки од ових кожних симптома, одмах се обратите лекару.
- Ако имате или сте имали нападе. Реците свом лекару што је пре могуће ако имате висок крвни притисак, главобољу, губитак вида.
- Ако имате дијабетес. Пацијенте са дијабетесом треба редовно проверавати ниво шећера у крви да би се утврдило да ли је потребно променити дозу лекова за дијабетес како би се смањио ризик од ниског шећера у крви.
Деца и адолесценти
Сутент није индициран за пацијенте млађе од 18 година. Сутент није проучаван код деце и адолесцената.
Интеракције Који лекови или храна могу да промене ефекат Сутента
Обавестите свог лекара или фармацеута ако узимате, недавно сте узимали или бисте могли узети било које друге лекове, укључујући и оне који се купују без рецепта и оне без рецепта.
Неки лекови могу променити ниво Сутента у организму. Требате рећи свом лекару ако узимате лекове који садрже следеће активне супстанце:
- кетоконазол, итраконазол - користи се за лечење гљивичних инфекција
- еритромицин, кларитромицин, рифампицин - користе се за лечење инфекција
- ритонавир - користи се за лечење СИДЕ
- дексаметазон - кортикостероид који се користи за неколико стања
- фенитоин, карбамазепин, фенобарбитал - користе се за лечење епилепсије и других неуролошких стања
- биљни препарати који садрже кантарион (Хиперицум перфоратум) - користе се за лечење депресије и анксиозности
Довољно са храном и пићем
Током лечења Сутентом треба избегавати унос сока од грејпа.
Упозорења Важно је знати да:
Трудноћа и дојење
Ако сте трудни или сумњате да сте трудни, обавестите свог лекара.
Сутент се не сме користити током трудноће осим ако је то крајње неопходно. Ваш лекар ће са вама разговарати о могућим ризицима примене Сутентног лечења током трудноће.
Ако је трудноћа могућа, морате користити поуздан метод контрацепције док се лечите Сутентом.
Ако дојите, реците то свом лекару. Не бисте требали дојити док се лечите Сутентом.
Вожња и управљање машинама
Ако се осећате вртоглаво или необично уморни, обратите посебну пажњу приликом вожње или рада са машинама.
Доза, начин и време примене Како се користи Сутент: Дозирање
Увек узимајте овај лек тачно онако како вам је рекао лекар.
Ако сте у недоумици, обратите се свом лекару. Ваш лекар ће вам прописати одговарајућу дозу, на основу врсте рака који морате да лечите. Ако се лечите од ГИСТ -а или МРЦЦ -а, уобичајена доза је 50 мг једном дневно која се узима 28 дана (4 недеље), након чега следи 14 дана (2 недеље) одмора (без лекова), у циклусима од 6 недеља.Ако се лечите од пНЕТ -а, уобичајена доза је 37,5 мг једном дневно, без одмора. Ваш лекар ће одредити потребну дозу и када треба прекинути Сутентни третман. Сутент се може узимати са или без хране.
Предозирање Шта треба учинити ако сте узели превише лека Сутент
Ако сте узели више лека Сутент него што је требало
Ако сте случајно узели превише капсула, одмах се обратите лекару. Можда ће бити потребна медицинска помоћ
Ако сте заборавили да узмете лек Сутент
Немојте узети двоструку дозу да бисте надокнадили заборављену дозу.
Нежељени ефекти Који су нежељени ефекти Сутент -а
Као и сви лекови, и овај лек може изазвати нежељена дејства, мада се она не морају јавити код свих.
Одмах се обратите лекару ако доживите било који од ових озбиљних нежељених ефеката (погледајте такође Шта треба да знате пре него што узмете лек Сутент):
Срчаних проблема. Реците свом лекару ако се осећате јако уморно, имате недостатак ваздуха или имате отечена стопала и глежњеве. То могу бити симптоми срчаних проблема, попут затајења срца и проблема са срчаним мишићима (кардиомиопатија).
Проблеми са плућима или дисањем. Реците свом лекару ако осетите кашаљ, бол у грудима, изненадну појаву недостатка ваздуха или искашљавање крви. То могу бити симптоми плућне емболије која се јавља када крвни угрушци путују до плућа.
Проблеми са бубрезима. Реците свом лекару ако осетите „измењену учесталост или одсуство мокрења, што би могли бити симптоми“ отказивања бубрега.
Крварење. Одмах обавестите свог лекара ако осетите било који од следећих симптома или озбиљан проблем са крварењем док узимате Сутент: отечени, болни стомак (стомак); повраћање са крвљу; тамне, лепљиве столице; главобоља или промене у менталном статусу, искашљавање крви или спутума са крвљу из плућа или дисајних путева.
Уништавање тумора који узрокује перфорацију црева. Реците свом лекару ако имате јаке болове у цревима, грозницу, мучнину, повраћање, крв у столици или промене у навикама црева.
Други нежељени ефекти који се могу јавити са Сутентом су:
Веома чести нежељени ефекти (могу се јавити код више од 1 на 10 особа)
- Смањење броја тромбоцита, црвених крвних зрнаца и / или белих крвних зрнаца (нпр. Неутрофила).
- Кратког даха
- Висок крвни притисак.
- Прекомерни умор, губитак снаге.
- Отицање узроковано течношћу испод коже и око очију, дубоким алергијским осипом.
- Бол / иритација у устима, бол / упала / сува уста, поремећаји укуса, узнемиреност желуца, мучнина, повраћање, пролив, затвор, бол у стомаку / отицање, губитак / смањење апетита.
- Смањена активност штитне жлезде (хипотироидизам).
- Вртоглавица
- Главобоља.
- Крварење из носа.
- Болови у леђима, болови у зглобовима.
- Бол у рукама и ногама.
- Пожутење коже / промена боје коже, прекомерна пигментација коже, промена боје косе, осип на длановима и табанима, осип, сува кожа.
- Кашаљ.
- Грозница.
- Потешкоће са заспањем.
Чести нежељени ефекти (могу се јавити код 1 до 10 на 100 људи)
- Формирање угрушака у крвним судовима.
- Недовољно снабдевање крви срчаном мишићу, због опструкције или сужења коронарних артерија.
- Бол у грудима.
- Смањена количина крви коју срце пумпа.
- Задржавање течности такође око плућа.
- Инфекције.
- Смањен ниво шећера у крви. Ако осетите знакове и симптоме ниског шећера у крви: Обавестите свог лекара што је пре могуће ако осетите умор, лупање срца, знојење, глад и губитак свести.
- Губитак протеина у урину, што понекад доводи до отока.
- Синдром грипе.
- Абнормални тестови крви, укључујући ниво ензима јетре и панкреаса.
- Висок ниво мокраћне киселине у крви.
- Хемороиди, ректални бол, крварење десни, отежано гутање или немогућност гутања.
- Печење или бол у језику, упала слузнице дигестивног тракта, вишак гасова у желуцу или цревима.
- Губитак тежине.
- Мишићно -коштани бол (бол у мишићима и костима), мишићна слабост, умор мишића, болови у мишићима, грчеви мишића.
- Сувоћа носа, зачепљеност носа.
- Прекомерно сузење.
- Промене у осетљивости коже, сува кожа, свраб, љуштење и упала коже, стварање пликова, акне, промена боје ноктију, губитак косе.
- Абнормалне сензације у екстремитетима.
- Прекомерно смањење / повећање осетљивости, посебно на додир.
- Спаљивање у стомаку.
- Дехидрација.
- Црвенило лица.
- Промена боје урина.
- Депресија.
- Зимица.
Мање чести нежељени ефекти (могу се јавити код 1 до 10 на 1.000 људи)
- Инфекције меких ткива, укључујући и аногениталну регију, потенцијално су опасне по живот. Одмах се обратите лекару ако осетите симптоме инфекције око ране на кожи, укључујући грозницу, бол, црвенило, отицање или дренажу гноја или крви.
- Удар.
- Срчани удар узрокован прекидом или смањеним дотоком крви у срце.
- Промене у електричној активности срца или измењени срчани ритам.
- Течност око срца (перикардни излив).
- Хепатична инсуфицијенција.
- Бол у стомаку (абдомену) узрокован „упалом панкреаса.
- Уништавање тумора узрокује перфорацију црева.
- Запаљење (отицање и црвенило) жучне кесе са или без камења.
- Абнормални комуникациони канал између две телесне шупљине или са кожом.
- Бол у устима, зубима и / или вилици, отицање или иритација у устима, утрнутост или осећај тежине у вилици или отпуштање зуба. Ово могу бити знаци и симптоми повреде кости вилице (остеонекроза). Одмах обавестите свог лекара и стоматолога ако приметите било који од ових знакова и симптома.
- Прекомерна производња хормона штитне жлезде доводи до повећаног метаболизма. Проблеми са зарастањем рана након операције.
- Повећање ензима мишића у крви (креатин фосфокиназа).
- Неодговарајућа и прекомерна реакција на алергене.
Ретки нежељени ефекти (могу се јавити код 1 до 10 на 10.000 људи)
- Тешке реакције коже и / или слузокоже (Стевенс-Јохнсонов синдром, токсична епидермална некролиза, мултиформни еритем).
- Синдром туморске лизе (ТЛС) - ТЛС обухвата скуп метаболичких компликација које могу настати током лечења карцинома. Узроковани су производима разградње захваћених ћелија рака и могу укључивати: мучнину, отежано дисање, неправилан рад срца, грчеве у мишићима, грчеве, замућен урин и умор повезан са абнормалним резултатима лабораторијских испитивања (висок ниво калијума, мокраћне киселине и фосфорне киселине) и низак ниво калцијума у крви) што може довести до промена у функцији бубрега и акутног затајења бубрега.
- Абнормални слом мишића који може узроковати бубрежне проблеме (рабдомиолиза).
- Оштећена функција мозга која може довести до различитих симптома, као што су главобоља, конфузија, напади и губитак вида (синдром задње реверзибилне леукоенцефалопатије).
- Болне улцерације на кожи (гангренозна пиодерма).
- Упала јетре (хепатитис).
- Запаљење штитне жлезде.
Пријављивање нежељених ефеката
Ако добијете било које нежељено дејство, обратите се свом лекару, што укључује и све могуће нуспојаве које нису наведене у овом упутству.Такође можете пријавити нежељена дејства директно путем националног система за пријављивање наведеног у Додатку В. Пријављивањем нежељених ефеката можете помоћи у пружању више информација о безбедности овог лека.
Истек и задржавање
- Чувајте овај лек ван погледа и дохвата деце.
- Немојте користити овај лек након истека рока употребе који је наведен на кутији и на етикети иза „ЕКСП“. Датум истека се односи на последњи дан у месецу.
- Овај лек не захтева посебне услове складиштења.
- Немојте користити овај лек ако приметите да је паковање оштећено или показује знаке неовлашћеног рада.
Не бацајте никакве лекове у отпадне воде или у кућни отпад. Питајте свог фармацеута како да баците лекове које више не користите. То ће помоћи заштити животне средине.
Шта Сутент садржи
Сутентне тврде капсуле од 12,5 мг
Активни састојак је сунитиниб. Свака капсула садржи сунитиниб малат еквивалентно 12,5 мг сунитиниба.
Остали састојци су:
- Садржај капсуле: манитол (Е421), натријум кроскармелоза, повидон (К-25) и магнезијум стеарат.
- Љуска капсуле: желатин, црвени гвожђе оксид (Е172) и титанијум диоксид (Е171).
- Тинта: шелак, пропилен гликол, натријум хидроксид, повидон и титанијум диоксид (Е171).
Сутентне тврде капсуле од 25 мг
Активни састојак је сунитиниб. Свака капсула садржи сунитиниб малат еквивалентно 25 мг сунитиниба.
Остали састојци су:
- Садржај капсуле: манитол (Е421), натријум кроскармелоза, повидон (К-25) и магнезијум стеарат.
- Љуска капсуле: желатин, титанијум диоксид (Е171), жути гвожђе оксид (Е172), црвени гвожђе оксид (Е172), црни гвожђе оксид (Е172).
- Тинта: шелак, пропилен гликол, натријум хидроксид, повидон и титанијум диоксид (Е171)
Сутентне тврде капсуле од 37,5 мг
Активни састојак је сунитиниб. Свака капсула садржи сунитиниб малат еквивалентно 37,5 мг сунитиниба.
Остали састојци су:
- Садржај капсуле: манитол (Е421), натријум кроскармелоза, повидон (К-25) и магнезијум стеарат.
- Овојница капсуле: желатина, титанијум диоксид (Е171), жути гвожђе оксид (Е172).
- Мастило: шелак, пропилен гликол, калијум хидроксид, црни гвожђе оксид (Е172).
Сутентне тврде капсуле од 50 мг
Активни састојак је сунитиниб. Свака капсула садржи сунитиниб малат еквивалентно 50 мг сунитиниба.
Остали састојци су:
- Садржај капсуле: манитол (Е421), натријум кроскармелоза, повидон (К-25) и магнезијум стеарат.
- Љуска капсуле: желатина, титанијум диоксид (Е171), жути гвожђе оксид (Е172), црвени гвожђе оксид (Е172) и црни гвожђе оксид (Е172).
- Тинта: шелак, пропилен гликол, натријум хидроксид, повидон и титанијум диоксид (Е171).
Опис како Сутент изгледа и садржај паковања
Сутент 12,5 мг је доступан у облику тврдих желатинских капсула са наранџастим поклопцем и телом, са "Пфизер" у белом мастилу на поклопцу и "СТН 12,5 мг" на телу, које садрже обојене грануле, жуто-наранџасте.
Сутент 25 мг је доступан у облику тврдих желатинских капсула са карамел капом и наранџастим телом, са "Пфизер" у белом мастилу на поклопцу и "СТН 25 мг" на телу, које садрже обојене грануле. Жуто-наранџасте.
Сутент 37,5 мг је доступан у облику тврдих желатинских капсула са жутим поклопцем и телом, са "Пфизер" у црном мастилу на поклопцу и "СТН 37,5 мг" на телу, које садржи обојене грануле, жуто-наранџасте.
Сутент 50 мг је доступан у облику тврдих желатинских капсула са чепом и телом боје карамеле, са "Пфизер" у белом мастилу на поклопцу и "СТН 50 мг" на телу, које садржи жуто-наранџасте грануле.. Доступан је у бочицама од 30 капсула и перфорираним блистерима за појединачну дозу који садрже 28 к 1 капсулу.
Не могу се на тржиште ставити све величине паковања.
Упутство о извору: АИФА (Италијанска агенција за лекове). Садржај објављен у јануару 2016. Присутне информације можда нису ажурне.
Да бисте имали приступ најновијој верзији, препоручљиво је да приступите веб страници АИФА (Италијанска агенција за лекове). Одрицање од одговорности и корисне информације.
01.0 НАЗИВ ЛИЈЕКА
СУТЕНТ 12,5 мг Тврди капсуле
02.0 КВАЛИТАТИВНИ И КВАНТИТАТИВНИ САСТАВ
Свака капсула садржи сунитиниб малат, што одговара 12,5 мг сунитиниба.
За потпуну листу помоћних супстанци погледајте одељак 6.1.
03.0 ФАРМАЦЕУТСКИ ОБЛИК
Тврда капсула.
Желатинске капсуле са наранџастим поклопцем и телом, означене белим мастилом „Пфизер“ на поклопцу, „СТН 12,5 мг“ на телу и садрже жуто-наранџасте грануле.
04.0 КЛИНИЧКЕ ИНФОРМАЦИЈЕ
04.1 Терапијске индикације
Стромални тумор гастроинтестиналног тракта (ГИСТ)
СУТЕНТ је индикован за лечење нересектабилног и / или метастатског гастроинтестиналног стромалног карцинома (ГИСТ) код одраслих особа након неуспешног лечења иматинибом због резистенције или нетолеранције.
Метастатски карцином бубрежних ћелија (МРЦЦ)
СУТЕНТ је индикован за лечење узнапредовалог / метастатског карцинома бубрежних ћелија (МРЦЦ) код одраслих.
Неуроендокрини тумори панкреаса (пНЕТ)
СУТЕНТ је индикован за лечење добро диференцираних, нересектабилних или метастатских неуроендокриних тумора панкреаса (пНЕТ) у прогресивној болести код одраслих.
Искуство са СУТЕНТ-ом као леком прве линије је ограничено (видети одељак 5.1).
04.2 Дозирање и начин примене
Терапију сунитинибом треба да започне лекар са искуством у примени лекова против рака.
Дозирање
За ГИСТ и МРЦЦ препоручена доза СУТЕНТ -а је 50 мг која се узима орално једном дневно, током 4 узастопне недеље, након чега следи 2 недеље одмора (распоред 4/2) како би се спровео цео курс од 6 недеља.
За пНЕТ, препоручена доза лека СУТЕНТ је 37,5 мг која се узима орално једном дневно, без заказаног периода одмора.
Прилагођавање дозе
Сигурност и подношљивост
За ГИСТ и МРЦЦ, дозе се могу мењати у корацима од 12,5 мг на основу индивидуалне безбедности пацијената и подношљивости. Дневна доза не би требало да пређе 75 мг нити да се смањи испод 25 мг.
За пНЕТ, дозе се могу мењати у корацима од 12,5 мг на основу индивидуалне безбедности и подношљивости пацијената. Максимална доза примењена у фази 3 пНЕТ студије била је 50 мг дневно.
Можда ће бити потребно обуставити унос неких доза на основу безбедности и подношљивости пацијента.
Инхибитори / индуктори ЦИП3А4
Треба избегавати истовремену примену сунитиниба са снажним индукторима ЦИП3А4, попут рифампицина (видети одељке 4.4. И 4.5). Ако то није могуће, можда ће бити потребно повећати дозу сунитиниба у корацима од 12,5 мг (до 87,5 мг / дан за ГИСТ и МРЦЦ или 62,5 мг / дан за пНЕТ) на основу пажљивог праћења толеранције.
Треба избегавати истовремену примену сунитиниба са моћним инхибиторима ЦИП3А4, попут кетоконазола (видети одељке 4.4 и 4.5). Ако то није могуће, можда ће бити потребно смањити дозу сунитиниба на минималну дозу од 37,5 мг дневно за ГИСТ и МРЦЦ или 25 мг дневно за пНЕТ, на основу пажљивог праћења толеранције.
Треба размотрити избор алтернативног истовременог лека без или са минималним потенцијалом да индукује или инхибира ЦИП3А4.
Посебне популације
Педијатријска популација
Безбедност и ефикасност сунитиниба код пацијената млађих од 18 година нису утврђени.
Нема доступних података.
Нема индикација за специфичну употребу сунитиниба код деце од рођења до 6 године у лечењу нересектабилног и / или метастатског ГИСТ -а након неуспеха лечења иматинибом због резистенције или нетолеранције. Нема индикација за специфичну употребу сунитиниба у педијатријској популацији у лечењу МРЦЦ-а и у лечењу добро диференцираних, нересектабилних или метастатских пНЕТ-а у прогресији болести.
Не препоручује се употреба сунитиниба у педијатријској популацији.
Старији пацијенти (≥ 65 година))
Отприлике једна трећина пацијената укључених у клиничка испитивања који су примали сунитиниб били су стари 65 година или старији. Нису примећене значајне разлике у безбедности и ефикасности између млађих и старијих испитаника.
Оштећење јетре
Није потребно прилагођавање почетне дозе када се сунитиниб даје пацијентима са благим или умереним оштећењем јетре (Цхилд-Пугх стадијум А и Б). Употреба сунитиниба код особа са тешким оштећењем јетре (Цхилд-Пугх стадијум Ц) није проучавана, па се не препоручује његова примена код пацијената са оштећењем јетре (видети одељак 5.2).
Оштећење бубрега
Није потребно прилагођавање почетне дозе када се сунитиниб примењује код пацијената са оштећењем бубрега (умерено до тешко) или бубрежном болешћу у завршном стадијуму (ЕСРД) на хемодијализи. Накнадно прилагођавање дозе треба извршити на основу безбедности и подношљивости пацијента (видети одељак 5.2).
Начин примене
СУТЕНТ је за оралну примену. Може се узимати са или без хране.
Ако се не узме доза, не треба давати додатну дозу. Пацијент треба да узме уобичајену прописану дозу следећег дана.
04.3 Контраиндикације
Преосетљивост на активну супстанцу или било коју помоћну супстанцу наведену у одељку 6.1.
04.4 Посебна упозорења и одговарајуће мере опреза при употреби
Треба избегавати истовремену примену са јаким индукторима ЦИП3А4 јер може смањити концентрацију сунитиниба у плазми (видети одељке 4.2 и 4.5).
Треба избегавати истовремену примену са моћним инхибиторима ЦИП3А4 јер може повећати концентрацију сунитиниба у плазми (видети одељке 4.2 и 4.5).
Поремећаји коже и ткива
Промјена боје коже, која може бити посљедица боје активне супстанце (жуте), врло је честа нежељена реакција која се јавља код приближно 30% пацијената. Пацијенте треба упозорити да током лијечења сунитинибом могу промијенити боју косе или могу се јавити и кожа. Остали могући дерматолошки ефекти могу укључивати сувоћу, задебљање или пуцање коже, пликове или повремени осип на длановима или табанима.
Горе наведене реакције нису биле кумулативне, генерално су биле реверзибилне и обично нису доводиле до прекида лечења. Пријављени су случајеви гангренозне пиодермије, генерално реверзибилне након престанка узимања лека. Пријављене су озбиљне кожне реакције, укључујући случајеве мултиформног еритема ( ЕМ) и случајеви који се могу приписати Стевенс-Јохнсоновом синдрому (СЈС) и токсичној епидермалној некролизи (НЕТ), неки од њих су фатални. Са знацима или симптомима СЈС, НЕТ или ЕМ (нпр. Сунитиниб треба прекинути. Ако се потврди дијагноза СЈС или НЕТ, лечење не треба настављати. У неким случајевима на сумњу на ЕМ, након повлачења реакције, пацијенти су поново увели терапију сунитинибом у толерисаним нижим дозама; неки од ових пацијената су такође примили истовремени третман кортикостероидима или антисептицима тамине.
Крварење и крварење узроковано туморима
Из искуства након стављања лијека у промет пријављени су хеморагични догађаји, неки са смртним исходом, укључујући гастроинтестинална, респираторна, уринарна и церебрална крварења.
Епизоде крварења су се јавиле код 18% пацијената лечених сунитинибом у поређењу са 17% пацијената у плацебо групи у студији ГИСТ фазе 3. Пацијенти са МРЦЦ на сунитинибу који никада раније нису били лечени сунитинибом пријавили су крварење у 39% случајева у поређењу са 11% пацијената лечених ИФН-α. Седамнаест (4,5%) пацијената на сунитинибу у поређењу са 5 (1,7%) пацијената на ИФН-α доживело је епизоде крварења степена 3 или више. Двадесет шест процената пацијената који су примали сунитиниб због МРЦЦ-а отпорног на цитокине пријавило је епизоде крварења. До крварења, искључујући епистаксу, дошло је код 21,7% пацијената лечених сунитинибом у поређењу са 9,85% пацијената у плацебо групи у фази 3 пНЕТ студије. Рутинска процена овог догађаја треба да укључи комплетну крвну слику и физички преглед.
Епистакса је била најчешћа нежељена хеморагијска реакција, пријављена је код приближно половине пацијената са солидним туморима који су пријавили крварење. Неке од ових епизода епистаксије биле су озбиљне, али врло ретко са смртним исходом.
Пријављени су догађаји крварења тумора, понекад повезани са некрозом тумора; неки од ових крварења били су фатални.
У клиничким испитивањима, "крварење тумора догодило се код приближно 2% пацијената са ГИСТ-ом. Ови догађаји се могу јавити изненада и, у случају рака плућа, могу се јавити у облику" тешке и по живот опасне хемоптизе или као плућна крварења. Случајеви плућног крварења, неки са смртним исходом, забележени су у клиничким испитивањима, а такође су пријављени и након стављања лека у промет код пацијената лечених сунитинибом за МРЦЦ, ГИСТ и рак плућа. СУТЕНТ није одобрен за употребу код пацијената са раком плућа.
Пацијенте који се истовремено лече антикоагулансима (нпр. Варфарином, аценокумаролом) може се периодично пратити комплетном крвном сликом (тромбоцити), факторима коагулације (ПТ / ИНР) и физичким прегледом.
Гастроинтестинални поремећаји
Пролив, мучнина / повраћање, бол у стомаку, диспепсија и стоматитис / орални бол били су најчешће пријављене гастроинтестиналне нежељене реакције; Пријављени су и случајеви езофагитиса (видети одељак 4.8).
Помоћна нега за гастроинтестиналне нежељене реакције које захтевају лечење могу укључивати лекове са антиеметичким, антидијарејским или антацидним својствима.
Озбиљне, понекад фаталне, гастроинтестиналне компликације, укључујући гастроинтестиналну перфорацију, догодиле су се код пацијената са интраабдоминалним малигнитетом леченим сунитинибом. У ГИСТ студији фазе 3, фатално гастроинтестинално крварење догодило се код 0,98% пацијената лечених плацебом.
Хипертензија
Хипертензија је била врло честа нежељена реакција пријављена у клиничким студијама. Доза сунитиниба је смањена или је привремено обустављена примена код приближно 2,7% пацијената код којих се појавила хипертензија. Ни код једног од ових пацијената сунитиниб није трајно прекинут. Тешка хипертензија (> 200 ммХг систолни или 110 ммХг дијастолни) десило се у 4,7% пацијената са солидним туморима.У МРЦЦ-у и претходно нелечених пацијената, случајеви хипертензије су пријављени у 33,9% пацијената на сунитинибу у поређењу са 3,6% пацијената на ИФН-α. тешка хипертензија се јавила код 12% претходно нелечених пацијената у групи сунитиниба и код мање од 1% пацијената из групе ИФН-α. У студији фазе 3 пНЕТ -а, хипертензија је пријављена код 26,5% пацијената који су узимали сунитиниб у поређењу са 4,9% пацијената у плацебо групи. Епизоде тешке хипертензије догодиле су се код пацијената са пНЕТ -ом у 10%. Пацијената који су лечени сунитинибом и 3% они који се лече плацебом. Пацијенте треба прегледати на хипертензију и на одговарајући начин их пратити. Привремена обустава лечења се препоручује код пацијената са тешком неконтролисаном хипертензијом уз лечење лековима. Лечење се може наставити када се хипертензија адекватно контролише.
Хематолошки поремећаји
Апсолутно смањење броја неутрофила 3. и 4. степена примећено је код 10%, односно 1,7% пацијената укључених у фазу 3 ГИСТ студије, односно код 16% и 1,6% пацијената укључених у студију. Фаза 3 МРЦЦ и код 13 % и 2,4% пацијената укључених у фазу 3 пНЕТ студије. Смањење броја тромбоцита 3 и 4 степена пријављено је код 3,7% односно 0,4% пацијената. Пацијенти укључени у фазу 3 ГИСТ студије, у 8,2% и 1,1% пацијената укључених у фазу 3 МРЦЦ студије и у 3,7% и 1,2% пацијената укључених у студију фазе 3 на пНЕТ.
Горе наведени догађаји нису били кумулативни, генерално су били реверзибилни и обично нису довели до прекида терапије. укључујући крварење повезано са тромбоцитопенијом и неутропеничним инфекцијама.
Уочено је да се анемија јавља у раним и касним фазама терапије сунитинибом; пријављени су степени 3 и 4.
Комплетна крвна слика треба да се уради на почетку сваког циклуса лечења код пацијената који примају сунитиниб.
Срчане патологије
Кардиоваскуларни догађаји, неки од њих са смртним исходом, укључујући затајење срца, кардиомиопатију, исхемију миокарда и инфаркт миокарда, пријављени су код пацијената лечених сунитинибом. Ови подаци указују да сунитиниб повећава ризик од кардиомиопатије. Код лечених пацијената нису идентификовани никакви специфични додатни фактори ризика за кардиомиопатију изазвану сунитинибом, осим специфичног дејства лека. Сунитиниб треба примењивати са опрезом код пацијената са ризиком од ових догађаја или који су већ имали такве догађаје.
У клиничким студијама, смањење ејекционе фракције леве коморе (ЛВЕФ) ≥ 20% и испод доње границе нормале догодило се у приближно 2% пацијената са ГИСТ-ом лечених сунитинибом, 4% пацијената са МРЦЦ-ом отпорним на цитокине лечене сунитинибом и у 2 % пацијената са ГИСТ-ом лечених плацебом. Чини се да ово смањење ЛВЕФ -а није прогресивно и често се побољшава наставком лечења. У студији спроведеној на пацијентима са МРЦЦ-ом који никада раније нису лечени, 27% пацијената лечених сунитинибом и 15% пацијената лечених ИФН-α имало је вредност ЛВЕФ испод доње границе нормале. За два пацијента (
Код пацијената са ГИСТ -ом, епизоде "срчане инсуфицијенције", "конгестивне срчане инсуфицијенције" или "инсуфицијенције леве коморе" пријављене су код 1,2% пацијената лечених сунитинибом и код 1% пацијената лечених плацебом. У кључној студији У фази 3 на ГИСТ-у (н = 312), фаталне срчане реакције повезане са лечењем су се јавиле код 1% пацијената у обе групе испитивања (сунитиниб и плацебо група). У студији друге фазе пацијената са цитокин-рефрактерним МРЦЦ-ом, 0,9 % пацијената је пријавило смртоносни инфаркт миокарда повезано са лечењем, а у фази 3 студије пацијената са МРЦЦ-ом који никада раније нису лечени, 0,6 % пацијената у руци ИФН-α и 0% пацијената у групи са сунитинибом пријавило је фаталне срчане догађаје. У фази 3 пНЕТ студије, један пацијент (1%) који је узимао сунитиниб имао је фаталну срчану инсуфицијенцију повезану са лечењем. Могућа корелација између инхибиције рецептора тирозин киназе (РТК) и срчане функције је нејасна.
Пацијенти који су пријавили срчане догађаје, у року од 12 месеци пре примене сунитиниба, као што су инфаркт миокарда (укључујући тешку / нестабилну ангину пекторис), операција коронарне / периферне премоснице, симптоматска ЦХФ, цереброваскуларни догађај или пролазни исхемијски напад или плућна емболија, били су искључени из клиничка испитивања са сунитинибом. Није познато да ли пацијенти са таквим истовременим стањима могу бити у повећаном ризику од развоја дисфункције леве коморе повезане са лековима.
Треба пажљиво пратити клиничке знакове и симптоме конгестивне срчане инсуфицијенције, посебно код пацијената са факторима ризика за срце и / или са историјом коронарне болести.
Лекарима се саветује да одмере овај ризик у односу на потенцијалне користи лека. Ове пацијенте треба пажљиво пратити због клиничких знакова и симптома конгестивне срчане инсуфицијенције током лечења сунитинибом.Основне и периодичне процене фракције избацивања леве коморе такође треба узети у обзир када се пацијент лечи сунитинибом. Код пацијената без срчаних фактора ризика, још увек треба размотрити процену вентрикуларне ејекционе фракције на почетку.
У присуству клиничких манифестација ЦХФ -а, препоручује се прекид терапије сунитинибом. Примену сунитиниба треба прекинути и / или смањити дозу код пацијената без клиничких доказа конгестивне срчане инсуфицијенције, али са смањењем избацивања између 20% и 50% од основне линије.
Продужење КТ интервала
Подаци из претклиничких студија (ин витро И ин виво), спроведене са дозама већим од оних које се препоручују код људи, указују на то да сунитиниб може инхибирати процесе срчане реполаризације (нпр. продужење КТ интервала).
До повећања КТц интервала на више од 500 мсец дошло је при стопи од 0,5%, а промене у односу на почетну вредност од више од 60 мсец догодиле су се у 1,1% од 450 пацијената са солидним туморима; оба ова параметра су препозната као потенцијално значајне варијације. У концентрацијама које одговарају приближно два пута терапијским концентрацијама, утврђено је да сунитиниб продужава интервал КТцФ (кориговано према Фредерициној формули).
Продужење КТ интервала проучавано је у студији која је обухватила 24 пацијента, у доби од 20-87 година, са узнапредовалим малигнитетима.Резултати ове студије показали су да је сунитиниб утицао на КТц интервал (дефинисан као плацебо прилагођена средња промена> 10 мсец, са горња граница од 90% ЦИ> 15 мсец) у терапијској концентрацији (3. дан) применом 24-часовне корекционе методе, а при концентрацијама изнад терапијске (9. дан) коришћењем обе корективне методе на почетку. Ниједан пацијент није пријавио вредност КТц> 500 мсец. Иако је ефекат на КТцФ интервал уочен 3. дана 24 сата након дозирања (тј. Са очекиваном терапијском концентрацијом у плазми након препоручене почетне дозе од 50 мг) помоћу 24-часовне корекционе методе, клинички значај овог налаза није јасан .
Користећи свеобухватне серијске процене ЕКГ -а које одговарају терапијским или експозицијама изнад терапијских, није примећен продужетак „озбиљног“ КТц интервала код ниједног пацијента у популацији која се може проценити или ИТТ (дакле једнака или већа од
Оцена 3 ЦТЦАЕ верзије 3.0).
При терапијским концентрацијама у плазми, средња максимална промена КТцФ интервала (коригована Фредерицином формулом) од почетне била је 9,6 мсец (90% ЦИ 15,1 мсец). При терапијским концентрацијама које одговарају приближно двоструко већим терапијским концентрацијама, максимална промена КТцФ интервала у односу на почетну вредност износио је 15,4 мсец (90% ЦИ 22,4 мсец).
Моксифлоксацин (400 мг) коришћен као позитивна контрола показао је средњу максималну промену КТцФ интервала у односу на почетну вредност од 5,6 мсек. Ниједан испитаник није пријавио ефекат на КТц интервал већи од степена 2 (ЦТЦАЕ верзија 3.0).
Продужење КТ интервала може узроковати повећан ризик од вентрикуларних аритмија, укључујући Торсаде де Поинтес.
Сунитиниб треба примењивати са опрезом код пацијената са историјом продужења КТ интервала, код пацијената лечених антиаритмицима, или са лековима који могу продужити КТ интервал, или код пацијената са већ постојећим релевантним срчаним обољењима, брадикардијом или абнормалностима. Електролитички. Истовремену примену сунитиниба са снажним инхибиторима ЦИП3А4 треба ограничити због могућег повећања концентрације сунитиниба у плазми (видети одељке 4.2 и 4.5).
Венски тромбоемболијски догађаји
Венски тромбоемболијски догађаји повезани са третманом примећени су у приближно 1,0% пацијената са солидним туморима леченим сунитинибом у клиничким студијама, укључујући студије ГИСТ и МРЦЦ.
У ГИСТ студији фазе 3, седам пацијената (3%) који су примали сунитиниб и ниједан пацијент у плацебо групи доживели су венске тромбоемболијске догађаје; пет од седам пацијената имало је дубоку венску тромбозу 3. степена (ДВТ), а два су имали дубоку венску тромбозу 1. или 2. Четворица од ових седам пацијената који су лечени од ГИСТ -а прекинути су након посматрања ДВТ -а.
Тринаест пацијената (3%) лечених сунитинибом у студији фазе 3 МРЦЦ-а, а никада раније нису лечени, а четири пацијента (2%) из две студије отпорне на цитокине МРЦЦ пријавило је венске тромбоемболијске догађаје. Девет од ових пацијената имало је венске тромбоемболијске догађаје. А " плућна емболија, један са степеном 2 и осам са степеном 4. Осам ових пацијената је имало ДВТ, један са степеном 1, два са степеном 2, четири са степеном 3 и један са степеном 4. Код пацијента са плућном емболијом у МРЦЦ студији , рефракторно на цитокине, доза је прекинута.
Од претходно нелечених пацијената са МРЦЦ-ом на ИФН-α, шест (2%) је пријавило венске тромбоемболијске догађаје, један пацијент (плућна емболија, сви степен 4).
Венски тромбоемболијски догађаји пријављени су за 1 (1,2%) пацијента у групи која је примала сунитиниб и 5 (6,1%) пацијената у групи која је примала плацебо у студији фазе 3 пНЕТ. Два од ових испитаника који су примали плацебо пријавили су ДВТ, један степена 2 и један степена 3.
У кључним студијама ГИСТ, МРЦЦ и пНЕТ нису забележени случајеви са фаталним исходом. Случајеви са фаталним исходом забележени су у постмаркетиншком окружењу производа (видети респираторне догађаје и одељак 4.8).
Артеријски тромбоемболијски догађаји
Забележени су случајеви артеријских тромбоемболијских догађаја (АТЕ), понекад фаталних, код пацијената лечених сунитинибом. Најчешћи догађаји укључивали су цереброваскуларну несрећу, пролазни исхемијски напад и церебралну исхемију. Фактори ризика повезани са артеријским тромбоемболијским догађајима, поред већ постојећег малигнитета и старости 65 година или више, укључивали су хипертензију, дијабетес мелитус и претходни тромбоемболијски догађај.
Респираторни догађаји
Пацијенти који су имали плућну емболију у претходних 12 месеци били су искључени из клиничких испитивања са сунитинибом.
Код пацијената који су примали сунитиниб у кључним студијама фазе 3, плућни догађаји (диспнеја, плеурални излив, плућна емболија или плућни едем) забележени су код приближно 17,8% пацијената са ГИСТ -ом, у приближно 26,7% оних са МРЦЦ -ом и у 12% од пацијенти са пНЕТ -ом.
Приближно 22,2% пацијената са солидним туморима, укључујући ГИСТ и МРЦЦ, лечено сунитинибом у клиничким студијама пријавило је плућне догађаје.
Случајеви плућне емболије забележени су код приближно 3,1% пацијената са ГИСТ -ом и приближно 1,2% пацијената са МРЦЦ -ом лечених сунитинибом у студијама фазе 3 (видети одељак 4.4. - Венски тромбоемболични догађаји). Нису примећени случајеви плућне емболије код пацијената са пНЕТ -ом третиран сунитинибом у студији фазе 3.Ретки случајеви са фаталним исходом забележени су у фази маркетинга производа (видети одељак 4.8).
Дисфункција штитне жлезде
Препоручује се процена функције штитне жлезде мерењем основних лабораторијских вредности код свих пацијената. Пацијенте са већ постојећим хипотироидизмом или хипертироидизмом треба лечити према стандардној клиничкој пракси пре почетка терапије сунитинибом. Током лечења сунитинибом, рутинску проверу функције штитне жлезде треба вршити свака 3 месеца. Осим тога, све пацијенте треба пажљиво пратити током лечења због могућих знакова и симптома дисфункције штитне жлезде, а пацијенте који развију било које знакове и / или симптоме који указују на дисфункцију штитне жлезде треба подвргнути лабораторијском испитивању функције штитне жлезде како се клинички очекује. Пацијенте код којих се развије дисфункција штитне жлезде треба лечити према стандардној клиничкој пракси.
Уочено је да се хипотироидизам јавља на почетку или на крају терапије сунитинибом.
Хипотироидизам је пријављен као нежељена реакција код 7 пацијената (4%) који су примали сунитиниб у две МРЦЦ студије спроведене на рефракторним пацијентима са цитокинима; код 61 пацијента (16%) који су примали сунитиниб и код три пацијента (
Додатно, повишење ТСХ-а је пријављено код 4 пацијента (2%) лечених од МРЦЦ-а отпорног на цитокине. Све у свему, 7% МРЦЦ популације пријавило је клиничке или лабораторијске доказе хипотиреозе повезане са лечењем. Стечени хипотироидизам је примећен код 6,2% пацијената у ГИСТ студији који су узимали сунитиниб у поређењу са 1% у плацебо групи. У фази 3 пНЕТ студије, хипотиреоза је пријављена код 6 пацијената (7,2%) који су примали сунитиниб и код једног пацијента (1,2%) који је примао плацебо.
Функција штитне жлезде је проспективно праћена у две студије код пацијената са раком дојке; СУТЕНТ није одобрен за лечење рака дојке. У једној студији, хипотироидизам је пријављен код 15 испитаника (13,6%) лечених сунитинибом и код 3 испитаника (2,9%) лечених стандардном терапијом. Повећан ТСХ у крви је забележен код 1 испитаника. (0,9%) лечених сунитинибом и ни код једног испитаника лечи стандардном терапијом. Хипертироидизам није забележен ни код једног испитаника леченог сунитинибом, а пријављен је код 1 испитаника (1,0%) који је примао стандардну терапију.У другој студији, хипотироидизам је пријављен код укупно 31 испитаника (13%) који су лечени сунитинибом и 2 испитаници (0,8%) лечени капецитабином Повећан ТСХ у крви забележен је код 12 испитаника (5,0%) лечених сунитинибом и ни код једног испитаника који је примао капецитабин. Хипертироидизам је забележен код 4 испитаника (1,7%) лечених сунитинибом и ни код једног испитаника који је лечен капецитабином. Смањени ТСХ у крви је забележен код 3 испитаника (1,3%) лечених сунитинибом и ни код једног испитаника. Лечен капецитабином. Повећање Т4 је забележено у 2 испитаника (0,8%) су лечени сунитинибом и 1 испитаник (0,4%) лечен капецитабином. Повећање Т3 пријављено је код 1 испитаника (0,8%) лечених сунитинибом и ни код једног испитаника који је лечен капецитабином.Сви пријављени догађаји повезани са штитном жлездом били су 1-2 степена.
Случајеви хипертиреозе, неки праћени хипотиреозом и случајеви тироидитиса ретко су пријављивани у клиничким испитивањима и у фази продаје производа.
Панкреатитис
Повећање активности липазе и амилазе у серуму примећено је код пацијената са неколико солидних тумора који су примали сунитиниб. Повећање активности липазе у серуму било је пролазно и генерално није било повезано са знацима и симптомима панкреатитиса код субјеката са солидним туморима различитих врста.
Панкреатитис је ретко примећен (
Пријављени су случајеви озбиљних догађаја панкреаса, неки са смртним исходом.
Ако се појаве симптоми панкреатитиса, терапију сунитинибом треба прекинути и пацијентима пружити одговарајућу подршку. У студији фазе 3 пНЕТ нису забележени догађаји повезани са лечењем панкреатитиса.
Хепатотоксичност
Хепатотоксичност је примећена код пацијената лечених сунитинибом. Случајеви затајења јетре, понекад са фаталним исходом, забележени су у функцији јетре (аланин трансаминаза [АЛТ], аспартат трансаминаза [АСТ], ниво билирубина). Знаци или симптоми затајења јетре, терапију сунитинибом треба прекинути и пацијентима пружити одговарајућу помоћну медицинску помоћ.
Хепатобилиарни поремећаји
Лечење сунитинибом може бити повезано са холециститисом, укључујући алитијазни холециститис и ентематозни холециститис. У кључним клиничким студијама учесталост холециститиса била је 0,5%.
Случајеви холециститиса забележени су у постмаркетиншком окружењу.
Бубрежна функција
Пријављени су случајеви оштећења бубрега, бубрежне инсуфицијенције и / или акутне бубрежне инсуфицијенције, у неким случајевима са смртним исходом.
Фактори ризика повезани са бубрежним оштећењем / отказивањем код пацијената који су примали сунитиниб су, поред већ постојећег карцинома бубрежних ћелија, укључивали: напредну старост, дијабетес мелитус, већ постојеће оштећење бубрега, затајење срца, хипертензију, сепсу, дехидратацију / хиповолемију и рабдомиолизу.
Безбедност наставка терапије сунитинибом код пацијената са умереном до тешком протеинуријом није систематски процењивана.
Пријављени су случајеви протеинурије и ретки случајеви нефротског синдрома. Препоручује се основна анализа урина и пацијенте треба пратити ради могућег развоја или погоршања протеинурије.
Лечење сунитинибом треба прекинути код пацијената са нефротским синдромом.
Фистуле
Ако дође до стварања фистуле, лечење сунитинибом треба прекинути. Постоје ограничени подаци о продуженом лечењу сунитинибом код пацијената са фистулама.
Погоршање процеса зарастања рана
За време терапије сунитинибом забележени су случајеви ослабљеног зарастања рана. Нису спроведене формалне клиничке студије о утицају сунитиниба на зарастање рана. Из разлога предострожности препоручује се привремена прекид терапије сунитинибом код пацијената који су подвргнути великој операцији. Клиничко искуство у погледу времена потребног за поновно покретање терапије након велике операције је ограничено. Стога се одлука о наставку терапије сунитинибом након велике операције мора темељити на клиничкој процјени опоравка од операције.
Остеонекроза мандибуле / максиле
Забележени су случајеви остеонекрозе вилице код пацијената лечених леком СУТЕНТ. У већини пријављених случајева, пацијенти су претходно или истовремено примали интравенозни третман бисфосфонатима, при чему је остеонекроза вилице идентификован ризик.Због тога треба бити опрезан када се СУТЕНТ и интравенозни бисфосфонати дају истовремено или узастопно.
Инвазивне стоматолошке интервенције су такође идентификоване као фактор ризика. Прије лијечења СУТЕНТ -ом треба размотрити стоматолошки преглед и одговарајућу превентивну стоматолошку његу. Ако је могуће, треба избегавати инвазивне стоматолошке интервенције код пацијената који су претходно узимали или узимају интравенозне бисфосфонате (видети одељак 4.8).
Преосетљивост / ангиоедем
Ако дође до едема услед реакције преосетљивости, лечење сунитинибом треба прекинути и дати стандардни медицински третман.
Поремећаји нервног система
Поремећаји укуса
Дисгеузија је пријављена код приближно 28% пацијената који су примали сунитиниб током клиничких испитивања.
Грчеви
У клиничким испитивањима са сунитинибом и након стављања лека у промет, примећени су случајеви напада код испитаника са или без радиолошких доказа о метастазама у мозгу. Осим тога, постојао је и ограничен број пријава (главобољу, смањену будност, оштећене менталне функције и губитак вида, укључујући кортикално слепило, треба контролисати медицинским третманом, укључујући контролу хипертензије. Препоручује се привремена суспензија сунитиниба; након повлачења у том случају, лечење се може наставити по нахођењу лекара који лечи.
Синдром туморске лизе (ТЛС)
Случајеви синдрома туморске лизе (ТЛС), неки са фаталним исходом, ретко су забележени у клиничким студијама и пријављени су након стављања лека у промет код пацијената лечених сунитинибом. Фактори ризика за ТЛС укључују велико оптерећење тумором, већ постојећу хроничну бубрежну инсуфицијенцију, олигурију, дехидратацију, хипотензију и кисели урин. Ове пацијенте треба пажљиво пратити и лечити према клиничким индикацијама; за ове пацијенте треба размотрити профилактичку хидратацију.
Инфекције
Пријављени су случајеви озбиљних инфекција, са или без неутропеније, укључујући и неке случајеве са фаталним исходом.
Инфекције које се најчешће примећују при лечењу сунитинибом су инфекције типичне за оболеле од рака, као што су респираторни, уринарни тракт, кожа и сепса.
Пријављени су ретки, понекад смртоносни случајеви некротизујућег фасциитиса, укључујући и перинеум. Терапију сунитинибом треба прекинути код пацијената код којих се развије некротизирајући фасциитис и одмах започети одговарајуће лечење.
Хипогликемија
Било је извештаја о смањењу глукозе у крви током лечења сунитинибом, неки од њих су клинички симптоматски и захтевају хоспитализацију због губитка свести. У случају симптоматске хипогликемије, терапију сунитинибом треба привремено прекинути. Ниво глукозе у крви треба редовно проверавати код пацијената са дијабетесом како би се проценило да ли је потребно прилагодити дозу лекова за дијабетес како би се смањио ризик од хипогликемије.
04.5 Интеракције са другим лековима и други облици интеракција
Студије интеракција су спроведене само код одраслих.
Лекови који могу повећати концентрације сунитиниба у плазми
Код здравих добровољаца, истовремена примена појединачне дозе сунитиниба са кетоконазолом, снажним инхибитором ЦИП3А4, довела је до повећања комбиноване Цмак [сунитиниб + примарни метаболит] Цмак и АУЦ0-∞ за 49%, односно 51%.
Примена сунитиниба са јаким инхибиторима ЦИП3А4 (нпр. Ритонавир) , итраконазол, еритромицин, кларитромицин, сок грејпа) могу повећати концентрацију сунитиниба.
Због тога треба избегавати комбинацију са инхибиторима ЦИП3А4 или размотрити алтернативни лек без или са минималним потенцијалом да инхибира ЦИП3А4.
Ако то није могуће, можда ће бити потребно смањити дозу СУТЕНТ -а на минимално 37,5 мг / дан за ГИСТ и МРЦЦ или 25 мг / дан за пНЕТ, на основу пажљивог праћења толеранције (видети одељак 4.2).
Лекови који могу смањити концентрације сунитиниба у плазми
Код здравих добровољаца, истовремена примена појединачне дозе сунитиниба и рифампицина индуктора ЦИП3А4 резултирала је смањењем Цмак [сунитиниб + примарни метаболит] Цмак и АУЦ0-∞ за 23% односно 46%.
Примена сунитиниба са јаким индукторима ЦИП3А4 (нпр. Дексаметазон, фенитоин, карбамазепин, рифампицин, фенобарбитал или биљни препарати који садрже кантарион)Хиперицум перфоратум) може смањити концентрацију сунитиниба. Стога треба избегавати повезаност са индукторима ЦИП3А4 или размотрити алтернативни лек који нема или има минималан потенцијал да индукује ЦИП3А4. Ако то није могуће, доза СУТЕНТ -а се може повећавати у корацима од 12, 5 мг (навише до 87,5 мг / дан за ГИСТ и МРЦЦ или 62,5 мг / дан за пНЕТ) на основу пажљивог праћења толеранције (видети одељак 4.2).
04.6 Трудноћа и дојење
Трудноћа
Нису спроведена испитивања на трудницама које су примале сунитиниб. Студије на животињама показале су репродуктивну токсичност, укључујући феталне малформације (видети одељак 5.3).
СУТЕНТ се не сме користити током трудноће или код жена које не користе ефикасне мере контрацепције, осим ако потенцијалне користи оправдавају потенцијални ризик за фетус. Ако се СУТЕНТ користи током трудноће, или ако пацијенткиња затрудне током лечења СУТЕНТ -ом, пацијенткињу треба обавестити о потенцијалним ризицима по фетус.
Женама у репродуктивном периоду треба саветовати да користе ефикасну контрацепцију и да избегавају трудноћу током лечења леком СУТЕНТ.
Време храњења
Сунитиниб и / или његови метаболити се излучују у млеко пацова. Није познато да ли се сунитиниб или његов главни активни метаболит излучују у мајчино млеко. Пошто се активне супстанце генерално излучују у мајчино млеко и с обзиром на могуће озбиљне нежељене реакције код дојенчади, жене не би требало да доје током лечења леком СУТЕНТ.
Плодност
Претклинички подаци указују на то да лечење сунитинибом може утицати на плодност мушкараца и жена (видети одељак 5.3).
04.7 Утицај на способност управљања возилима и машинама
Нису спроведена испитивања утицаја на способност управљања возилима и рада са машинама.Пацијенте треба обавестити о могућој појави вртоглавице током терапије сунитинибом.
04.8 Нежељени ефекти
Сажетак сигурносног профила
Најозбиљније нежељене реакције повезане са лечењем сунитинибом, неке са смрћу, су бубрежна инсуфицијенција, срчана инсуфицијенција, плућна емболија, перфорација гастроинтестиналног тракта и крварења (нпр.
Најчешће нежељене реакције било ког степена (о којима су извештавали пацијенти у кључним клиничким испитивањима за РЦЦ, ГИСТ и пНЕТ) укључивале су смањење апетита, поремећаје укуса, хипертензију, умор, гастроинтестиналне сметње (нпр. Пролив, мучнина, стоматитис, диспепсија и повраћање), промену боје синдрома коже и палмарно-плантарне еритродисестезије. Ови симптоми се могу смањити наставком лечења. Хипотироидизам може настати током лечења. Хематолошки поремећаји (нпр. Неутропенија, тромбоцитопенија и анемија) били су међу најчешћим нежељеним реакцијама на лекове.
Смртоносни нежељени догађаји осим оних наведених у одељку 4.4. или у одељку 4.8 и сматра се да је могуће повезано са сунитинибом, укључујући отказивање више органа, дисеминирану интраваскуларну коагулацију, перитонеално крварење, надбубрежну инсуфицијенцију, пнеумоторакс, шок и изненадну смрт.
Табела нежељених реакција
Нежељене реакције које су пријавили пацијенти са ГИСТ, МРЦЦ и пНЕТ у обједињеном скупу података од 7115 пацијената наведене су доле и категорисане према органским системима, учесталости и озбиљности (НЦИ-ЦТЦАЕ). Пријављене су и постмаркетиншке нежељене реакције идентификоване у клиничким студијама. Унутар сваке класе учесталости, нежељени ефекти се пријављују према падајућој озбиљности.
Учесталости су дефинисане на следећи начин: врло честе (≥1 / 10), уобичајене (≥1 / 100а
Табела 1 - Нежељене реакције пријављене у клиничким студијама
Следећи појмови су груписани:
назофарингитис и орални херпес
б Бронхитис, инфекција доњих дисајних путева, упала плућа и инфекција респираторног тракта
ц Апсцес, апсцес удова, анални апсцес, апсцес десни, апсцес јетре, апсцес панкреаса, перинеални апсцес, периректални апсцес, ректални апсцес, поткожни апсцес и зубни апсцес
д Кандидиаза једњака и орална кандидијаза
и целулитис и инфекција коже
ф Сепса и септички шок
г Абдоминални апсцес, абдоминална сепса, дивертикулитис и остеомијелитис
х Смањен апетит и анорексија
и Дисгеузија, агеузија и промена укуса
ј Акутни коронарни синдром, ангина пекторис, нестабилна ангина, оклузија коронарне артерије, исхемија миокарда
к Смањење / аномалија фракције избацивања
л Акутни инфаркт миокарда, инфаркт миокарда, тихи инфаркт миокарда
м Орофарингеални и фаринголарингеални бол
н Стоматитис и афтозни стоматитис
о Бол у стомаку, бол у доњем и горњем делу трбуха
п Гастроинтестинална и цревна перфорација
к Холециститис и алитски холециститис
р Жута кожа, промена боје коже и поремећаји пигментације
с псоријазиформни дерматитис, ексфолијативни осип, осип, еритематозни осип, фоликуларни осип, генерализовани осип, макуларни осип, макуло-папуларни осип, папуларни осип и пруритицни осип
т Реакција коже и патологија коже
у Патологија ноктију и промена боје ноктију
в Умор и астенија
в Едем лица, едем и периферни едем
к амилаза и повећана амилаза
* Укључује фаталне догађаје
Опис идентификованих нежељених реакција
Инфекције и инфестације: Пријављени су случајеви озбиљних инфекција (са или без неутропеније), укључујући случајеве са смртним исходом. Пријављени су случајеви некротизујућег фасциитиса, понекад са смрћу, који би такође могао утицати на подручје перинеума (видети такође одељак 4.4).
Поремећаји крви и лимфног система: Пријављени су случајеви тромботичне микроангиопатије. У овим случајевима препоручује се привремена суспензија СУТЕНТ -а; након решавања ових догађаја, лечење се може поново започети по нахођењу лекара који лечи.
Поремећаји имунолошког система: Пријављене су реакције преосјетљивости, укључујући ангиоедем.
Поремећаји нервног система: Пријављено је неколико случајева, неки са смртним исходом, код особа које су имале нападе и радиолошке доказе реверзибилног синдрома задње леукоенцефалопатије (РПЛС) (видети такође одељак 4.4).
Поремећаји метаболизма и исхране: Пријављена је већа инциденца хипогликемијских догађаја код пацијената са пНЕТ у поређењу са пацијентима са МРЦЦ и ГИСТ. Међутим, сматрало се да многи од нежељених догађаја уочених у клиничким испитивањима нису повезани са испитиваним третманом.
Хепатобилијарни поремећаји: Пријављена је дисфункција јетре и може укључивати абнормалности тестова функције јетре, хепатитис или отказивање јетре.
Поремећаји коже и поткожног ткива: Пријављени су случајеви гангренозне пиодерме, генерално реверзибилне након престанка лечења (видети такође одељак 4.4).
Поремећаји мишићно -коштаног система и везивног ткива: Пријављени су случајеви миопатије и / или рабдомиолизе, неки повезани са акутном бубрежном инсуфицијенцијом. Пацијенте са знацима или симптомима мишићне токсичности треба лечити у складу са стандардном медицинском праксом.
Пријављени су случајеви стварања фистуле, понекад повезани са туморском некрозом и регресијом, у неким случајевима са фаталним исходом.
Забележени су случајеви остеонекрозе вилице код пацијената лечених леком СУТЕНТ, од којих су се многи јављали код пацијената који су препознали факторе ризика за остеонекрозу вилице, нарочито изложеност интравенозним бисфосфонатима и / или историју зубних болести које захтевају инвазивне стоматолошке интервенције (види такође одељак 4.4).
Пријављивање сумње на нежељене реакције
Извештавање о сумњи на нежељене реакције које се јаве након добијања дозволе за лек важно је јер омогућава континуирано праћење односа користи и ризика лека. Од здравствених радника се тражи да пријаве све сумње на нежељене реакције путем националног система за пријављивање. У „Анексу В .
04.9 Предозирање
Не постоји специфичан противотров за предозирање сунитинибом, а лечење предозирања треба да укључи опште мере подршке. Ако је назначено, уклањање неапсорбоване активне супстанце може се постићи повраћањем или испирањем желуца. Пријављени су. Случајеви предозирања; у неким од у овим случајевима нежељене реакције које су се јавиле биле су у складу са познатим безбедносним профилом сунитиниба.
05.0 ФАРМАКОЛОШКА СВОЈСТВА
05.1 Фармакодинамичка својства
Фармакотерапијска група: антинеопластични агенси, инхибитори протеин киназе.
АТЦ код: ЛО1КСЕ04.
Механизам дејства
Сунитиниб инхибира рецепторе више тирозин киназе (РТК) који су укључени у раст тумора, неоангиогенезу тумора и метастатско напредовање рака. Сунитиниб је идентификован као инхибитор рецептора фактора раста изведених из тромбоцита (ПДГФРα и ПДГФРβ), рецептора васкуларног ендотелног фактора раста (ВЕГФР1, ВЕГФР2 и ВЕГФР3), рецептора фактора матичних ћелија (КИТ), рецептора тирозин киназе ФЛТ3 (Фмс-лике тирозин киназа 3), ЦСФ-1Р рецептор (рецептор фактора стимулације колоније (ЦСФ-1Р)) и рецептор неутрофног фактора изведеног из глија (РЕТ). Главни метаболит показује моћ упоредиву са сунитинибом у биохемијским и ћелијским тестовима.
Клиничка ефикасност и безбедност
Безбедност и клиничка ефикасност сунитиниба проучавани су у лечењу пацијената са ГИСТ -ом резистентним на иматиниб (тј. Оних пацијената који су напредовали током или после лечења иматинибом) или нетолерантни на иматиниб (тј. Они који су имали значајну токсичност током лечења иматинибом) који је спречио наставак лечења), у лечењу пацијената са МРЦЦ -ом и у лечењу пацијената са неоперабилним пНЕТ -ом.
Ефикасност се заснива на времену до прогресије тумора и повећању преживљавања код пацијената са ГИСТ-ом, преживљавању без прогресије и објективној стопи одговора код претходно нелечених пацијената са МРЦЦ и МРЦЦ, рефракторним на терапију са МРЦЦ, цитокинима, и на преживљавању без прогресије болести са пНЕТ -ом.
Стромални тумори гастроинтестиналног тракта (ГИСТ)
Почетна отворена студија ескалације спроведена је код пацијената са ГИСТ-ом након неуспеха иматиниба (средња максимална дневна доза 800 мг) због резистенције или нетолеранције. Уписано је 97 пацијената са различитим дозама и режимима дозирања; 55 пацијената је лечено СУТЕНТ-ом 50 мг према препорученом четворонедељном леку и двонедељном распореду повлачења лека (распоред 4/2).
У овој студији, просечно време до прогресије ТТП је било 34,0 недеље (95% ЦИ = 22,0-46,0 недеља).
Случајна, двоструко слепа, плацебом контролисана студија фазе 3 сунитиниба спроведена је код пацијената са ГИСТ-ом који су били нетолерантни или су имали прогресију болести током или након лечења иматинибом (средња максимална дневна доза 800 мг). У овој студији, 312 пацијената (2: 1) је рандомизовано да се лечи са 50 мг сунитиниба или плацеба, орално једном дневно према распореду 4/2 до прогресије болести или повлачења из студије из „другог разлога (207 пацијената је примило сунитиниб и 105 плацеба).
Примарна крајња тачка ефикасности студије била је ТТП, дефинисана као време од рандомизације до прве документације о објективној прогресији тумора.У време унапред одређене међу-анализе, средњи ТТП са сунитинибом био је 28,9 недеља (95% ЦИ = 21,3-34,1 недеља) ) када га је испитао истраживач, и 27,3 недеље (95% ЦИ = 16,0-32,1 недеља) када га је проценио Независни одбор за ревизију и статистички је био супериорнији у односу на 5,1-недељни ТТП добијен са плацебом (95% ЦИ = 4,4-10,1 недеље, п
Независна комисија за ревизију. Разлика у укупном преживљавању била је статистички у корист сунитиниба [Размера опасности: 0,491 95% (ЦИ 0,290-0,831)]; ризик од смрти био је 2 пута већи код пацијената у групи која је примала плацебо него у онима у групи која је примала сунитиниб.
Након привремене анализе ефикасности и безбедности, на препоруку независног ДСМБ-а, студија је заслепљена, а пацијентима у плацебо групи понуђено је да пређу на отворену терапију сунитинибом.
Укупно 255 пацијената је лечено сунитинибом у отвореној фази лечења у студији, укључујући 99 пацијената који су у почетку лечени плацебом.
Анализа примарних и секундарних крајњих тачака у отвореној фази студије поново је потврдила резултате добијене у време привремене анализе, као што је приказано у доњој табели.
Табела 2 - Преглед крајњих тачака ефикасности (ИТТ популација)
Средње укупно преживљавање (ОС) у популацији ИТТ износило је 72,7 недеља и 64,9 недеља (ХР 0,876, 95% ЦИ 0,679 - 1.129, п = 0,306) у групи која је примала сунитиниб, односно у групи која је примала плацебо. У овој анализи, плацебо група укључивала је оне пацијенте који су рандомизирани на плацебо, а затим су прешли на отворену терапију сунитинибом.
Метастатски карцином бубрежних ћелија (МРЦЦ) код претходно нелечених пацијената
Рандомизована, мултицентрична, међународна студија фазе 3 спроведена је да би се проценила ефикасност и безбедност сунитиниба у поређењу са интерфероном ИФН-α код претходно нелечених пацијената са МРЦЦ-ом. Седамсто педесет пацијената је рандомизовано 1: 1 у групе за лечење; понављајући шестонедељни циклуси. Сваки циклус се састоји од 4 недеље са 50 мг дневно орално, након чега следе 2 недеље без узимања лека (распоред 4/2), или са ИФН-α који се примењује орално поткожно у дози од 3 милиона јединица (МУ ) прве недеље, 6 МУ друге недеље и од треће недеље надаље у дози од 9 МУ према третману од 3 узастопна дана сваке недеље.
Просечно трајање лечења било је 11,1 месеци (распон: 0,4 - 46,1) за лечење сунитинибом и 4,1 месеца (распон 0,1 - 45,6) за лечење ИФН -α. Озбиљни нежељени догађаји повезани са третманом (ТРСАЕ) пријављени су код 23,7% пацијената који су примали сунитиниб и 6,9% пацијената који су примали ИФН-α. Међутим, стопе прекида због нежељених догађаја биле су 20% за сунитиниб и 23% за ИФН-α. Прекид лијечења догодио се код 202 пацијента (54%) у групи која је примала сунитиниб и 141 пацијента (39%) у групи која је примала ИФН-α.
До смањења дозе дошло је код 194 пацијената (52%) лечених сунитинибом и 98 пацијената (27%) лечених ИФН-α. Пацијенти су се лечили до прогресије болести или повлачења студије. Примарна крајња тачка ефикасности било је преживљавање без прогресије (ПФС).
Планирана привремена анализа показала је статистички значајну предност за СУТЕНТ у односу на ИФН-α. У овој студији, средњи ПФС за групу сунитиниба био је 47,3 недеље у поређењу са 22,0 недеље за групу ИФН-α; однос опасности је био 0,415 (95% ЦИ: 0,320-0,539, п-вредност
Лечење сунитинибом било је повезано са дужим преживљавањем од третмана ИФН-α. Просечно укупно преживљавање износило је 114,6 недеља за групу са сунитинибом (95% ЦИ: 100,1 - 142,9 недеља) и 94,9 недеља за групу са ИФН -α (95% ЦИ: 77,7 - 117,0 недеље) са Размера опасности од 0,821 (95% ЦИ: 0,673-1,001; п = 0,0510 на основу нестратификованог лог-ранк теста).
Преживљавање без прогресије (ПФС) и укупно преживљавање (ОС), посматрано у намери лечења (ИТТ) популације и одређено радиолошком лабораторијском проценом, сумирани су у следећим табелама:
Сажетак крајњих тачака ефикасности (ИТТ популација)
Цитокин-рефракторни метастатски карцином бубрежних ћелија (МРЦЦ)
Студија фазе 2 спроведена је са СУТЕНТ-ом код пацијената који су били отпорни на претходну терапију цитокинима лечених интерлеукин-2 или ИФН-α. Шездесет и три пацијента су примала оралну почетну дозу од 50 мг сунитиниба једном дневно током 4 узастопне недеље, након чега је уследио двонедељни одмор да би се завршио цео 6-недељни курс (распоред лечења 4/2). Примарна крајња тачка ефикасности била је објективна стопа одговора (објективан
Стопа Одговора (ОРР)) према критеријумима РЕЦИСТ (Критеријуми за процену одговора у солидним туморима).
У овој студији, објективна стопа одговора била је 36,5% (95% ЦИ 24,7% -49,6%), а средње време до прогресије (ТТП) било је 37,7 недеља (95% ЦИ 24,0-46,4 недеље).
Отворена, једнокрака, мултицентрична, потврдна студија за процену ефикасности и безбедности сунитиниба спроведена је код пацијената са МРЦЦ рефракторним на претходну терапију цитокинима. Сто и шест пацијената је примило најмање једну дозу од 50 сунитиниба у мг. оквир шеме 4/2.
Примарна крајња тачка ефикасности ове студије била је стопа ОРР -а, док су секундарне крајње тачке укључивале ТТП, трајање одговора (ДР) и укупно преживљавање (ОС).
У овој студији, ОРР је био 35,8% (95% ЦИ 26,8% -47,5%). ДР и медијан ОС још нису достигнути.
Неуроендокрини тумори панкреаса (пНЕТ)
Отворена, мултицентрична, студија подршке у фази 2 процењивала је ефикасност и безбедност монотерапије сунитинибом у дневним дозама од 50 мг према распореду 4/2 [4 недеље лечења, 2 недеље паузе] код пацијената са неоперабилним пНЕТ-ом У кохорти од 66 код пацијената са карциномом ћелија оточића панкреаса, крајња тачка примарног одговора била је 17%.
Спроведена је кључна фаза 3, мултицентрична, међународна, рандомизована, двоструко слепа, плацебом контролисана студија монотерапије сунитинибом код пацијената са неоперабилним пНЕТ-ом.
Пацијенти за које је требало да су документовали прогресију болести засновану на РЕЦИСТ-у у претходних 12 месеци били су рандомизовани (1: 1) да примају 37,5 мг сунитиниба једном дневно без заказаног периода каренце (н = 86) или плацебо (н = 85) .
Примарни крајњи циљ била је процена преживљавања без прогресије (ПФС) код пацијената који су узимали сунитиниб у односу на оне који су примали плацебо. Остали крајњи циљеви били су ОС, проценат ОРР, исходи пријављени од пацијената) и безбедност.
Са становишта демографских карактеристика, групе пацијената лечених сунитинибом и плацебом биле су упоредиве. Осим тога, 49% пацијената који су примали сунитиниб и 52% пацијената који су примали плацебо имали су нефункционалне туморе, а 92% пацијената на обе руке имало је метастазе у јетри. Студија је дозволила употребу аналога соматостатина. 66% пацијената лечених сунитинибом и 72% пацијената лечених плацебом претходно је лечено системском терапијом. Осим тога, 24% пацијената у групи сунитиниба и 22% пацијената који су примали плацебо група је примала аналоге соматостатина. Клинички значајна предност сунитиниб ПФС -а у односу на плацебо примећена је при процени истраживача. Средња вредност ПФС -а била је 11,4 месеца у групи која је примала сунитиниб у поређењу са 5, 5 месеци у групи која је примала плацебо [однос ризика: 0,418 (95% ЦИ 0,263 , 0,662), п-вредност = 0,0001]; слични резултати су примећени када су процене одговора тумора, засноване на примени РЕЦИСТ-а на мерењима тумора које су извршили истраживачи, коришћене за одређивање прогресије болести као што је приказано у Табели 3. А Размера опасности у корист сунитиниба је примећен у свим подгрупама за које су процењене основне карактеристике, укључујући анализу према броју претходних системских терапија. Укупно 29 пацијената у групи са сунитинибом и 24 у плацебо групи није добило претходну системску терапију; код ових пацијената није било "Размера опасности за ПФС било је 0,365 (95% ЦИ 0,156, 0,857), п = 0,0156.
Слично, међу 57 пацијената у групи која је примала сунитиниб (укључујући 28 са 1 претходном системском терапијом и 29 са 2 или више системских терапија) и 61 пацијент у плацебо групи (укључујући 25 са 1 претходном системском терапијом и 36 са 2 или више системских терапија ) л "Размера опасности за ПФС било је 0,456 (95% ЦИ 0,264, 0,787), п = 0,0036.
Анализа осетљивости ПФС -а изведена је када је ПФС заснован на истраживачким мерењима тумора и када су сви субјекти, цензурисани из других разлога осим завршетка студије, сматрани догађајима ПФС -а. Ова анализа је дала конзервативну процену ефекта третмана сунитинибом и потврдила примарну анализу, показујући „Размера опасности 0,507 (95% ЦИ 0,350, 0,733), п = 0,000193. Кључна студија у НЕТ -у панкреаса је прерано прекинута на основу препоруке независног одбора за процену лекова, а примарна крајња тачка заснована је на процени истраживача: оба стања су могла утицати на процену ефекта лечења.
Да би се искључила пристрасност у процени истражитеља ПФС -а, спроведен је независни, слепи централни преглед дијагностичког снимања; овај преглед је подржао процену истраживача, као што је приказано у Табели 3.
Табела 3 - Резултати ефикасности из фазе 3 пНЕТ студије
ЦИ = интервал поверења, ХР = Однос опасности, НА = Није применљиво, НР = није достигнуто
двострани не-стратификовани тест лог-ранга
б Фисхеров егзактни тест
Укупна цифра преживљавања није била зрела када је спроведена анализа. Било је 9 смртних случајева у групи која је примала сунитиниб, а 21 у плацебо групи. Уочена је статистички значајна разлика у објективној стопи одговора на сунитиниб у поређењу са плацебом.
У случајевима прогресије болести, пацијенти су обавештени о лечењу које су примали, а пацијентима који су узимали плацебо понуђена је могућност да буду укључени у другу отворену продужену студију са сунитинибом. Због раног затварања студије, преостали пацијенти су информисани о лечењу које су примали и понуђен им је приступ отвореној продуженој студији са сунитинибом. Укупно 59 пацијената у групи која је примала плацебо лечено је сунитинибом у студији. продужетка.
Резултати Упитника о квалитету живота Европске организације за истраживање и лечење рака (ЕОРТЦ КЛК-Ц30) показали су да је укупан квалитет живота повезан са здрављем и пет функционалних домена (физичко, уложно, когнитивно, емоционално и социјални) били су конзервирани код пацијената који су примали сунитиниб, у поређењу са онима који су примали плацебо, са мало симптоматских нуспојава.
Педијатријска популација
Европска агенција за лекове одложила је обавезу достављања резултата студија са СУТЕНТ -ом у једној или више подскупина педијатријске популације за лечење ГИСТ -а (видети одељак 4.2 за информације о педијатријској употреби).
Европска агенција за лекове одустала је од обавезе достављања резултата студија са СУТЕНТ -ом у свим подгрупама педијатријске популације за лечење бубрежних и бубрежних карличних карцинома (искључујући нефробластом, нефробластоматозу, чисте ћелије саркома, мезобластичну нефрому, бубрежну медуларну жлезду) карцином и рабдоидни тумор бубрега) (видети одељак 4.2 за информације о педијатријској употреби).
Европска агенција за лекове одустала је од обавезе достављања резултата студија са СУТЕНТ -ом у свим подскупинама педијатријске популације за лечење гастроентеропанкреатичних неуроендокриних тумора (са искључењем неуробластома, неуроганглиобластома, феохромоцитома) (видети одељак 4.2 за информације о педијатријској употреби) ).
05.2 "Фармакокинетичка својства
Фармакокинетика сунитиниба процењена је код 135 здравих добровољаца и код 266 пацијената са солидним туморима. Фармакокинетика је била слична у свим тестираним популацијама солидних тумора и у здравих добровољаца.
У оквиру режима дозирања од 25-100 мг, површина испод криве (АУЦ) и Цмак се повећавају пропорционално дози. Уз поновљену дневну примену, сунитиниб се акумулира 3-4 пута, а главни активни метаболит акумулира 7-10 пута. Равнотежне концентрације сунитиниба и његовог главног активног метаболита постижу се у року од 10-14 дана. До 14. дана, комбиноване концентрације сунитиниба у плазми и његовог главног активног метаболита су 62,9 - 101 нг / мЛ и представљају предвиђене циљне концентрације засноване на претклиничким подацима за инхибицију фосфорилације рецептора. ин витро и довести до смањења застоја / раста тумора ин виво.
Главни активни метаболит чини 23-37% укупне изложености леку. Нису примећене значајне промене у фармакокинетици сунитиниба или главног активног метаболита при понављању једном дневно или поновљеним курсевима у тестираним режимима дозирања.
Апсорпција
Након оралне примене сунитиниба, највеће концентрације (Цмак) се генерално примећују унутар 6-12 сати (Тмак) од уноса лека.
Храна нема утицаја на биорасположивост сунитиниба.
Дистрибуција
Везивање сунитиниба и његовог главног активног метаболита за протеине хумане плазме у тестовима у витро износили су 95% односно 90%, без "очигледне зависности од концентрације.
Привидни волумен дистрибуције (Вд) сунитиниба био је велики - 2.230 л - што указује на дистрибуцију у ткивима.
Метаболичке интеракције
Израчунате вредности Ки ин витро за све испитиване изоформе цитокрома (ЦИП) (ЦИП1А2, ЦИП2А6, ЦИП2Б6, ЦИП2Ц8, ЦИП2Ц9, ЦИП2Ц19, ЦИП2Д6, ЦИП2Е1, ЦИП3А4 / 5 и ЦИП4А9 / 11) показало се да је мало вероватно да ће сунитиниб и његов главни активни метаболит бити а метаболизам других активних супстанци које се могу метаболизовати помоћу ових ензима.
Биотрансформација
Сунитиниб се примарно метаболише помоћу ЦИП3А4, изоформе цитокрома П450, који производи његов главни активни метаболит, десетил сунитиниб, који се даље метаболише помоћу истог изоензима.
Треба избегавати истовремену примену сунитиниба и јаких индуктора или инхибитора ЦИП3А4 јер се нивои сунитиниба у плазми могу променити (видети одељке 4.4 и 4.5).
Елиминација
Излучивање се углавном одвија путем измета (61%), а бубрежна елиминација непромењене активне супстанце и њених метаболита представља 16% примењене дозе. Сунитиниб и његов главни активни метаболит били су главна једињења идентификована у плазми, урину и измету и чинили су 91,5%, 86,4%и 73,8%респективно радиоактивности откривене у обједињеним узорцима. Мањи метаболити су идентификовани у урину и измету, али генерално нису откривени у плазми. Укупни орални клиренс (ЦЛ / Ф) износио је 34-62 Л / х.
Након оралне примене код здравих добровољаца, полувреме елиминације сунитиниба и његовог главног активног метаболита десетила је приближно 40-60 сати, односно 80-110 сати.
Посебне популације
Оштећење јетре: Сунитиниб и његов главни метаболит се примарно метаболишу у јетри. Системска изложеност након појединачне дозе сунитиниба била је слична код субјеката са благим или умереним оштећењем јетре (Цхилд-Пугх фазе А и Б) у поређењу са субјектима са нормалном функцијом јетре. СУТЕНТ није проучаван код испитаника са тешким оштећењем јетре (Цхилд-Пугх фаза Ц).
Студије карцинома су искључиле пацијенте са АЛТ или АСТ> 2,5 к ГГН (горња граница нормале) или ако су ове вредности биле> 5,0 к ГГН због метастаза у јетри.
Оштећење бубрега: Фармакокинетичке анализе становништва показале су да на очигледни клиренс сунитиниба (ЦЛ / Ф) није утицао клиренс креатинина у разматраном распону (42-347 мл / мин). Системска изложеност након једне појединачне дозе сунитиниба била је слична код особа са тешким оштећењем бубрега ( ЦЛцр80 мл / мин). Иако сунитиниб и његов главни метаболит нису уклоњени хемодијализом код испитаника са ЕСРД, укупна системска изложеност била је 47% нижа за сунитиниб и 31% за његов главни метаболит у поређењу са субјектима са нормалном бубрежном функцијом.
Тежина, статус перформанси: Фармакокинетичке анализе становништва указују да нису потребне прилагодбе почетне дозе за тежину и статус перформанси Источне задружне онколошке групе (ЕЦОГ).
Пол припадности: Доступни подаци указују на то да женке могу имати 30% мањи привидни клиренс сунитиниба (ЦЛ / Ф) од мушкараца; ова разлика, међутим, не захтева прилагођавање почетне дозе.
05.3 Предклинички подаци о безбедности
Студије токсичности при поновљеним дозама код пацова и мајмуна у трајању до 9 месеци показале су да су главни ефекти на циљне органе у гастроинтестиналном тракту (повраћање и дијареја код мајмуна); надбубрежне жлезде (кортикална конгестија и / или крварење код пацова и мајмуна, са некрозом праћеном фиброзом код пацова); крв и лимфни систем (хипоцелуларност коштане сржи и лимфоидно исцрпљивање тимуса, слезине и лимфних чворова); егзокрини панкреас (дегранулација ацинарних ћелија са једноћелијском некрозом); пљувачне жлезде (ацинарна хипертрофија); коштани зглобови (задебљање плоче за раст); материце (атрофија) и јајника (смањење развоја фоликула). Сви резултати су се догодили са клинички значајним нивоима изложености сунитиниба у плазми. Остали ефекти уочени у студијама су: продужење КТц интервала, редукција избацивања леве коморе (ЛВЕФ) и тестиса тубуларна атрофија, повећање мезангијалних ћелија у бубрегу, крварење у гастроинтестиналном тракту и оралној слузници и хипертрофија ћелија предње хипофизе. ендометријума) и плоча за раст костију (задебљање епифизе или дисплазија хрскавице) повезани су са фармаколошким дејством сунитиниба. Већина ових догађаја била је реверзибилна након прекида терапије током 2-6 недеља.
Генотоксичност
Генотоксични потенцијал сунитиниба је процењен ин витро и ин виво. Сунитиниб није показао мутагене ефекте на бактерије применом метаболичке активације коју је обезбедила јетра пацова. Сунитиниб није изазвао структурне хромозомске аберације у периферним лимфоцитима људи ин витро. Полиплоидија (аберације у броју хромозома) примећена је у периферним лимфоцитима људи у витро, и у присуству и у одсуству метаболичке активације. Сунитиниб није показао кластогени потенцијал у коштаној сржи пацова ин виво. Главни активни метаболит није процењен на генотоксичан потенцијал.
Карциногеност
У једномесечној студији да би се пронашао распон доза (0, 10, 25, 75 или 200 мг / кг / дан) у којима су расХ2 трансгени мишеви храњени оралном сондом дозирањем дневно и континуирано дозом када се даје, на примећене су највеће дозе (200 мг / кг / дан) карцинома и хиперплазија Брунерових жлезда дуоденума.
Проведена је шестомјесечна студија за процјену карциногености код расХ2 трансгених мишева храњених оралном епруветом дозирањем примијењене дозе дневно (0, 8, 25, 75 [смањено на 50] мг / кг / дан). У дозама ≥25 мг / кг / дан примењеним током 1 или 6 месеци (≥7,3 пута већа од АУЦ -а виђена код пацијената који су примали препоручену дневну дозу [РДД]), гастродуоденалних карцинома, примећена је повећана учесталост у позадини. Хемангиосарком и / или хиперплазија слузнице желуца.
У двогодишњој студији за процену канцерогености код пацова (0, 0,33, 1 или 3 мг / кг / дан) када се сунитиниб примењивао у 28-дневним циклусима, након чега су уследили 7-дневни периоди без примене, утврђено је повећање инциденце феохромоцитом и медуларна хиперплазија надбубрежне жлезде код мушких пацова који су примали дозу од 3 мг / кг / дан више од 1 године (≥ 7,8 пута већа од АУЦ код пацијената који су примали РДД). Карцином Брунерове жлезде јавио се у дуоденуму у дозама ≥ 1 мг / кг / дан код жена, у дозама од 3 мг / кг / дан код мушкараца, а хиперплазија ћелија слузокоже била је евидентна у желучаним жлездама у једнаким дозама. дан код мушкараца; ови догађаји су се догодили у дозама од 0,9, 7,8 и 7,8 пута од АУЦ код пацијената који су примали РДД. Релевантност ових неопластичних налаза уочених у студијама карциногености миша (расХ2) и пацова за људе лечене сунитинибом је неизвесна.
Токсичност по репродукцију и развој
У студијама о репродуктивној токсичности нису уочени ефекти на плодност мушкараца или жена. Међутим, студије токсичности при поновљеним дозама спроведене на пацовима и мајмунима показале су ефекте на плодност женки у облику фоликуларне атрезије, дегенерације жутог тела, ендометријалних промена у материци и смањења тежине материце и јајника до нивоа клинички значајног системско излагање. Ефекти на плодност мужјака пацова примећени су у облику тубуларне атрофије у тестисима, смањења сперматозоида у епидидимису и колоидног исцрпљивања у простати и семенским мехурићима при нивоима изложености плазми 25 пута већој од системске изложености људи.
Код пацова, ембрионално-фетални морталитет резултирао је значајним смањењем броја живих фетуса, повећаним бројем ресорпција, повећаним постимплантацијским губицима и укупним губитком потомака у 8 од 28 трудних женки при нивоима изложености плазми 5,5 пута системској изложености код људи . Код зечева је смањење тежине трудне материце и броја живих фетуса узроковано повећањем ресорпције фетуса, губицима након имплантације и укупним губитком потомства код 4 од 6 трудних женки при троструким нивоима изложености плазми. Системска изложеност код људи. Третман сунитинибом код пацова током органогенезе резултирао је развојним ефектима при дозама ≥ 5 мг / кг / дан, који се састоје од повећане учесталости феталних скелетних малформација, које углавном карактерише одложено окоштавање торакалних / лумбалних пршљенова и јављају се при нивоима изложености плазми 5.5 пута системске изложености човека. Код зечева, развојни ефекти састојали су се од повећане учесталости расцепа усне при нивоима изложености плазми приближно једнаким онима који су примећени у клиници, и расцепа усне и непца при нивоима изложености плазми 2,7 пута од системске изложености код човека.
Ефекти сунитиниба (0,3; 1,0; 3,0 мг / кг / дан) на пре- и постнатални развој процењени су у студији на трудним женкама пацова. Повећање телесне тежине мајке током гестације и лактације смањено је при дозама> 1 мг / кг / дан, али није примећена репродуктивна токсичност мајке до доза од 3 мг / кг / дан (процењена изложеност> 2,3 пута већа од АУЦ -а нађене код пацијената) смањена телесна тежина потомака примећена је у дозама од 3 мг / кг / дан у периодима пре и после одбијања. Није примећена развојна токсичност у дозама од 1 мг / кг / дан (изложеност приближно ≥0,9 пута већа од АУЦ виђена код пацијената који примају РДД).
06.0 ФАРМАЦЕУТСКЕ ИНФОРМАЦИЈЕ
06.1 Помоћне супстанце
Садржај капсуле
Манитол (Е421)
Натријум кроскармелоза
Повидон (К-25)
Магнезијум стеарат
Љуска капсуле
Јелли
Црвени оксид гвожђа (Е172)
Титанијум диоксид (Е171)
Инк
Шелак
Пропилен гликол
Натријум хидроксид
Повидоне
Титанијум диоксид (Е171)
06.2 Некомпатибилност
Није битно.
06.3 Период важења
3 године.
06.4 Посебне мере предострожности при складиштењу
Овај лек не захтева посебне услове складиштења.
06.5 Природа непосредног паковања и садржај паковања
Полиетиленске боце велике густине (ХДПЕ), са затварачем од полипропилена, које садрже 30 тврдих капсула.
Прозирни мехурићи од поли (хлоротрифлуороетилена) / ПВЦ -а са перфорираним јединичним дозама и са алуминијумском фолијом са топлотно запечаћеним лаком који садржи 28 к 1 тврде капсуле.
Не могу се на тржиште ставити све величине паковања.
06.6 Упутства за употребу и руковање
Нема посебних упутстава.
07.0 НОСИЛАЦ ОВЛАШЋЕЊА ЗА ПРОМЕТ
Пфизер Лтд
Рамсгате Роад
Сендвич, Кент ЦТ13 9Њ
УК
08.0 БРОЈ ОДЛИКЕ ЗА ПРОМЕТ
ЕУ/1/06/347/001
037192022
ЕУ/1/06/347/004
09.0 ДАТУМ ПРВОГ ОДОБРЕЊА ИЛИ ОБНОВЕ ОВЛАШЋЕЊА
Датум прве ауторизације: 19. јул 2006
Датум последњег обнављања: 09. јануар 2012
10.0 ДАТУМ РЕВИЗИЈЕ ТЕКСТА
11.0 ЗА РАДИО ДРОГЕ, ПОТПУНИ ПОДАЦИ О ДОСИМЕТРИЈИ ИНТЕРНОГ ЗРАЧЕЊА
12.0 ЗА РАДИО -ЛИЈЕКОВЕ, ДОДАТНА ДЕТАЉНА УПУТСТВА О ПРИМЕРНОЈ ПРИПРЕМИ И КОНТРОЛИ КВАЛИТЕТА