Наследно стање, СМА је последица мутација у гену СМН1 или гену СМН2, чија је сврха да произведе протеин који служи за осигуравање опстанка моторних неурона.
Постоји пет различитих облика спиналне мишићне атрофије: тип 0, тип 1, тип 2, тип 3 и тип 4. Прва три типа су веома озбиљна и узрокују превремену смрт пацијента; тип 3 и тип 4 су блаже варијанте, које утичу на животни стандард пацијента, али без изазивања преране смрти.
За дијагностицирање СМА потребан је генетски тест на узорку крви.
Тренутно се терапија СМА углавном заснива на симптоматским третманима, чији је циљ ублажавање поремећаја и контрола компликација. Доступан је лек, заснован на принципима генске терапије, али то је веома скупо решење и применљиво само на одређене пацијенте.
, што се манифестује атрофијом и последичним слабљењем скелетних мишића, те моторичким тешкоћама.
СМА је стање које може изазвати смрт пацијента у младој или веома младој доби: најозбиљнији облици болести, у ствари, утичу на ефикасност респираторних мишића и одговорни су за епизоде респираторне инсуфицијенције или упале плућа са фатални исход.
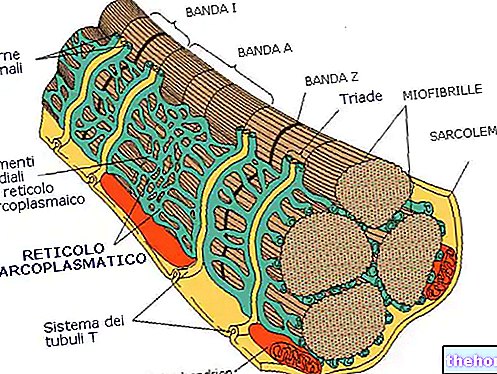
Моторни неурони и СМА

Моторни неурони или моторни неурони су нервне ћелије које настају у централном нервном систему (мозак и кичмена мождина) и које помоћу својих продужетака (аксони) контролишу активност мишића и жлезда.
Постоје две врсте моторних неурона: горњи моторни неурони (или први моторни неурони) и доњи моторни неурони (или други моторни неурони).
Горњи моторни неурони потичу из мозга и усмеравају активност доњих моторних неурона, који настају углавном у кичменој мождини и одговорни су за усмеравање активности скелетних (или соматских) мишића, глатких (или висцералних) мишића, срчаног мишића и срца.
Моторни неурони појединаца са СМА постепено се дегенерирају, узрокујући „мишићну атрофију због неактивности која у најтежим случајевима доводи до парализе, респираторне инсуфицијенције и смрти.
Епидемиологија: колико је честа спинална мишићна атрофија?
СМА има „годишњу учесталост од 1 случаја на 10 000 новорођених.
5 и од којих зависи производња такозваног протеина преживљавања моторних неурона (СМН).Као што назив протеина који производе СМН1 и СМН2 сугерише, мутација ових гена лишава моторне неуроне биолошке супстанце неопходне за њихов опстанак; тачније, смањује ниво протеина: на пример, у присуству мутација у СМН1, нивои СМН протеина падају на 10-20% нормалног.
Очигледно, одсуство одговарајућих количина СМН протеина одређује прогресивну дегенерацију моторних неурона.
Губитак моторних неурона прекида живчану сигнализацију која омогућава контролу активности мишића људског тијела; потоњи, као посљедица чињенице да више нису за употребу, пролазе постепени процес атрофије и слабљења.
Да ли сте знали да ...
Ген СМН2 је, за СМА, ген који модификује болест; у ствари, код пацијената са мутацијом у СМН1 и који из неког разлога имају три или четири копије гена СМН2, СМА се јавља у блажем облику.
Спинална мишићна атрофија: типови мутација
Када је СМА последица "промене СМН1, у 95-98% случајева одговорна мутација се састоји у брисању целог гена, док се само у 2-5% у" аномалији нормалне секвенце гена.
Спинална мишићна атрофија: наследна болест
У готово свим случајевима (98%), генетска аномалија одговорна за СМА је наследна, односно преносе је родитељи болесне јединке.
2% ненаследних случајева СМА је последица мутације де ново настао у врло раној фази ембрионалног развоја.
СМА и модел наслеђивања
Модел наслеђивања за спиналну мишићну атрофију је аутосомно рецесиван, што значи да је за наслеђивање СМА неопходно да су оба родитеља здрави носиоци генетског дефекта у СМН1 или СМН2 и да га оба родитеља преносе.
У случају аутосомно рецесивних наследних болести као што је СМА, вероватноћа да ће оба здрава носиоца пренети генетски дефект на дете и тако се разболети је 25%или један у свака 4 случаја.
Врсте СМА
На основу старости почетка и озбиљности стања, стручњаци препознају пет различитих облика спиналне мишићне атрофије:
- СМА тип 0: то је најтежи облик од свих. Манифестује се и пре рођења са смањеном покретљивошћу плода.
Бебе обично преживе неколико недеља након рођења, чак и када добију респираторну подршку. - СМА тип 1: од облика који се јављају током живота, он је најтежи и најчешћи (око 50% случајева); појављује се у раном добу, обично у шестом месецу живота.
По правилу, то је узрок смрти већ у првим годинама живота; ретко, током адолесценције.
Смрт се обично јавља због „респираторне инсуфицијенције“ или инфекције плућа. - СМА тип 2: то је облик који гравитацијом заузима друго место; генерално, почиње између 7 и 18 месеци живота.
Очекивани животни век за погођене је већи него у претходном случају: пацијенти, заправо, успевају да достигну пунолетство. - СМА тип 3: мање озбиљан од претходна два, овај облик СМА се типично јавља након 18 месеци живота (у неким случајевима може се појавити и током детињства или адолесценције).
Укључује велике инвалидности, али не утиче на очекивани животни век. - СМА тип 4: то је одрасли облик болести, као и најтежи; обично почиње око треће деценије живота и има веома спор ток.
Генерално није одговоран за респираторне проблеме и повезан је са „нормалним очекиваним животним веком“.
Нивои СМН протеина утичу на тежину СМА: што је мања количина СМН, већа је озбиљност повезане болести.
Смањење нивоа СМН уско је повезано са обимом генетског дефекта који је утицао на гене СМН1 или СМН2: што је овај дефект опсежнији, то је значајније смањење количине СМН протеина (то је случај, нпр. на пример, брисање гена).
Надаље, СМА не угрожава интелектуалне функције (ИК пацијената је нормалан) и штеди орган вида.
За додатне информације: СМА: сви симптомиСимптоми СМА типа 0
Као што је раније речено, тип 0 СМА се већ јавља у пренаталном добу са смањеном покретљивошћу плода; при рођењу, дакле, болесно дете има евидентне потешкоће у гутању и дисању.
Болест доводи до смрти у року од неколико недеља од рођења, чак и када пацијент добије респираторну подршку.
СМА симптоми типа 1
Деца са СМА тип 1 имају веома слабе мишиће који се не развијају како би требало (трошење мишића). Ово их спречава у обављању активности као што су подизање главе, померање удова и заузимање седећег положаја; штавише, поступно компликује виталне функције, попут сисања млека, гутања, жвакања и дисања.
Типично, СМА типа 1 је фатална у првих неколико година живота; неки пацијенти, међутим, успевају да достигну адолесценцију.
Смрт се обично јавља услед респираторне инсуфицијенције или „инфекције плућа услед потешкоћа са гутањем (гутање или упала плућа) аб ингестис).
Симптоми СМА типа 2
СМА тип 2 се класично манифестује са:
- Мекоћа мишића руку и ногу;
- Дрхтање у прстима и рукама;
- Потешкоће у самосталном заузимању седећег положаја (међутим, пацијент успева да га одржи);
- Потешкоће при стајању и ходању
- Деформитети и проблеми са зглобовима;
- Отежано дисање и гутање хране;
- Сколиоза (обично се јавља касније).
Чак и у овој ситуацији, отежано дисање и гутање хране узрок су преране смрти, која се обично јавља на почетку одрасле доби.
Симптоми СМА типа 3
СМА тип 3 изазива проблеме са држањем и равнотежом, дрхтањем руку и потешкоћама при устајању из сједећег положаја, ходању, пењању уз степенице и трчању.
У почетку болести не захтијевају подршку за кретање; касније, с дегенерацијом већег броја моторних неурона, штаке, ходалице и инвалидска колица постају темељне.
Иако се то може догодити, врло је ријетко да пацијенти са СМА тип 3 пате од проблема с дисањем и гутањем хране.
У присуству овог облика СМА, очекивани животни век је нормалан, али са свим горе наведеним проблемима.
Симптоми СМА типа 4
Са почетком одраслих, СМА тип 4 се обично повезује са:
- Слабљење мишићног тонуса у рукама и ногама;
- Потешкоће при ходању
- Дрхтање и нагло трзање мишића.
У почетку су горе наведене притужбе умерене; у старости постају доследнији.
Као и СМА типа 3, СМА типа 4 није болест која би утицала на очекивани животни век пацијента.
СМА: када видети доктора?
Свим родитељима који знају да су здрави носиоци СМА препоручује се консултација са педијатром са искуством у генетским болестима и генетичаром.
Ако немате информације ове врсте, добро је из месеца у месец проценити моторички развој вашег детета и функције од којих живот зависи (нпр. Дисање).
Свакако, немогућност седења или заузимања седећег положаја, потешкоће у храњењу, присуство респираторних дефицита и танка и мање напета мускулатура од оне код вршњака представљају звона за узбуну.
Што се тиче СМА за одрасле, сумња се на мање или више изненадну појаву мишићне слабости и отежано ходање и треба их пратити.
Спинална мишићна атрофија: Компликације
Најтежи облици СМА могу довести до компликација као што су:
- Гушење од хране. То је због смањене способности жвакања и уноса хране.
- Респираторна инсуфицијенција. То је последица немогућности контроле активности респираторних мишића.
- Упала плућа аб ингестис (или инхалациона пнеумонија). Јавља се када страни материјал који преноси патогене, попут хране, пљувачке или назалног секрета, уђе или се накупља у плућима.
Упала плућа аб ингестис резултат је тешкоћа при гутању. - Парализа која је резултирала употребом инвалидских колица. То се дешава када је болест непоправљиво угрозила пацијентове локомоторне способности.
- Неухрањеност. То је још једна последица тешкоћа при гутању: пацијент се, у ствари, бори да се правилно храни.
Треба напоменути да се понекад током дијагнозе СМА могу користити тестови попут електромиографије или биопсије мишића.
СМА: Физички преглед и анамнеза
Физички преглед код пацијента који може патити од СМА укључује пажљиву анализу симптома и тражење неких типичних знакова болести, као што су:
- Слабост и осетљивост мишића;
- Нагле контракције мишића
- Смањени или одсутни тетивни рефлекси.
Што се тиче историје болести, ово се углавном фокусира на породичну историју пацијента, како би се утврдило да ли се било који други члан породице (родитељи, браћа и сестре, бака и деда) жале или су се жалили на сличну симптоматологију. Очигледно, чињеница да је СМА наследна болест, пренета од родитеља.
Иако не дозвољавају постављање коначне дијагнозе, физички преглед и историја болести могу пружити врло корисне информације које усмеравају истраге ка спровођењу генетског теста.
Јасно је да ако је пацијент мало дете, родитељи ће током историје болести комуницирати са лекаром.
СМА и генетски тест

Генетски тест за откривање СМА укључује претраживање и проучавање мутација у генима СМН1 / СМН2 у узорку крвних зрнаца пацијента.
Присуство генетских промена очигледно значи болест.
Анализа откривених мутација је неопходна за утврђивање врсте присутне спиналне мишићне атрофије и озбиљности стања.
Да би се сазнали резултати горе поменутог генетског теста, генерално је потребно сачекати 3 до 4 недеље (прецизно време чекања варира у зависности од генетског центра који спроводи тест).
СМА: да ли је могућа пренатална дијагноза?
Могуће је дијагностиковати СМА у пренаталном добу.
Да бисте то урадили, потребан вам је генетски тест на узорку феталних ћелија, добијен деликатним методама, попут вилоцентезе или амниоцентезе.
С обзиром на ризик од побачаја који карактерише ЦВС и амниоцентезу, лекари се баве пренаталним истраживањем било каквих мутација приписаних „спиналној мишићној атрофији само ако иза тога стоји породична историја СМА или ако је нерођено дете дете здравих носилаца болести.
СМА и скрининг новорођенчади
Треба напоменути да је у неколико италијанских региона (Лацио и Тоскана) услуга активна скрининг за рану дијагнозу СМА и других озбиљних генетских болести.
Рана дијагноза ових болести омогућава благовремено планирање најприкладније симптоматске терапије за контролу симптома и компликација.
Спинална мишићна атрофија и планирање трудноће
Генетско саветовање се препоручује свим женама које траже трудноћу које:
- Имали су дете са СМА у претходној трудноћи;
- Иза себе имају породичну историју СМА;
- Да ли су здрави преносиоци болести или им је партнер.
Генетско саветовање може помоћи женама са овим стањима да схвате каквим је ризицима изложено будуће дете.
СМА и диференцијална дијагноза
Постоје две патологије врло сличне СМА, које само "темељна дијагностичка истрага препознаје и спречава забуну са" спиналном мишићном атрофијом: то су "спинална мишићна атрофија са респираторним дистресом (СМАРД) и" булбо-спинална мишићна атрофија (БСМА). Или Кеннедијева болест); први је последица мутације гена ИГХМБП2 који се налази на хромозому 11, док је други последица мутације полног хромозома Кс.
и фармацеутски производи) одобрила Золгенсму, методу генске терапије за лечење спиналне мишићне атрофије.
Золгенсма се састоји од високо напредне технике молекуларне биологије, која укључује употребу вектора вируса способног да убаци нормалну копију гена СМН1 / СМН2 у ДНК присутну у моторним неуронима пацијента.
Примена горе поменутог вектора вируса се врши интравенозном ињекцијом.
Золгенсма се показала ефикасном. Међутим, како се очекивало, он има два главна ограничења која спречавају његову уобичајену употребу:
- Веома је скупо. Говори се о милионима евра;
- Примењује се само на СМА пацијенте млађе од 2 године.
Спинална мишићна атрофија: симптоматски третмани
Симптоматска терапија за СМА гарантује веће користи ако се одмах усвоји; ово чини рану дијагнозу болести веома важном.
СМА и респираторна подршка
Одговарајућа респираторна подршка помаже пацијентима са СМА не само да дишу, већ и смањују ризик од инфекција плућа.
Међу различитим терапеутским могућностима, постоје маске за неинвазивну вентилацију и инвазивнија рјешења попут оротрахеалне интубације и трахеостомије; прва су идеална за мање тешке случајеве, док су инвазивнија рјешења неопходна за пацијенте с озбиљним проблемима.
СМА и подршка у исхрани
Најтежи облици спиналне мишићне атрофије утичу на способност гутања и жвакања хране, излажући пацијента ризику од гушења, гутања упале плућа и потхрањености.
За сузбијање ових опасних последица, неопходно је прибећи помоћним средствима за храњење, као што је назогастрична сонда или операција гастростомије, и ослонити се на нутриционисту који ће планирати дијету прилагођену потребама пацијента.
СМА и физиотерапија
Моторне потешкоће које карактеришу пацијента са спиналном мишићном атрофијом доводе до укочености зглобова и мишића услед неактивности.
Одговарајући програм физиотерапије омогућава вам да, колико је то могуће, побољшате флексибилност мишића и учините зглобове мање крутим.
Јасно је да овај програм укључује вежбе чије извођење је надохват могућности пацијента.
СМА и ортопедија
У присуству сколиозе, типичне за тешке облике СМА, неопходно је консултовати ортопеда; ово друго би могло указивати на употребу ортопедског корзета, ако је деформација блага, или на операцију спиналне фузије, ако је кичмена малформација озбиљна.
Лекови против СМА
Неколико година постоје и специфични лекови против СМА.
Ови лекови заслужују посебан третман у поређењу са симптоматским терапијама, иако не дозвољавају излечење болести, већ само да је обуздају.
Специфични лекови против СМА који су тренутно доступни су Спинраза (нусинерсен) и Еврисди (рисдиплам): први делују тако што исправљају аберантну производњу протеина СМН у процесу; други повећава нивое производње СМН, такође покушавајући да их задржи на квота адекватна потребама људског организма.
Одобрени од ФДА 2017. и 2020. године, Спинраза и Еврисди гарантују резултате, у неким случајевима чак и више него задовољавајуће, међутим имају важно ограничење: веома су скупи.
За додатне информације: Спинраза: Како функционише, ризици и користи